- 4192 Devonwood Way, Ashburn, Virginia, 20148
- Helpline: +1 (703) 665-3747
qPCR is not PCR Just as a Straightjacket is not a Jacket-the Truth Revealed by SARS-CoV-2 False-Positive Test Results
- Home
- Back to Journal
- Article Details
Abstract
RT-qPCR for molecular detection of SARS-CoV-2 in human respiratory tract specimens is inherently flawed. It is a fluorophore-labeled probe technology based on ssDNA hybridization, using PCR as a tool to determine if the intended molecular hybridization has indeed taken place by measuring the momentum of PCR-dependent probe degradation, which is expressed as Ct value, a point number selected from the curve of fluorescence signal accumulation during PCR cycling. The technology dictates that the nucleotide sequences of the two PCR primers and the probe must be fully matched with the sequences of their corresponding partners on the target DNA template. However, identification of three conserved regions within a short region of the virus genome, typically <100 bases, may not always be possible. In theory all PCR primers and probes may fail to detect the common mutants of SARS-CoV-2 when high stringency PCR conditions are used. Lowering the PCR stringency may lead to amplification of nontarget DNA and favors nonspecific hybridization, resulting in false positives. Routine sequencing of a 300–400 bp cDNA PCR amplicon is a method for definitive detection of SARS-CoV-2. For primer design, it is easy to locate two 21-base sequences, which are conserved and flank an intended template of 300 to 400 bases in length for DNA sequencing. Emerging virus variants with interprimer nucleotide changes, deletions or insertions do not escape sequencing detection. Single nucleotide mutations at the primer-binding sites usually do not cause failure of PCR amplification of an intended template. PCR is a tool invented to provide templates for DNA sequencing analysis. The fluorescence signal generated in RT-qPCR is not a reliable surrogate marker for the DNA sequence of a target template.
Keywords: SARS-CoV-2; False-positive; False-negative; PCR; RT-PCR; RT-qPCR; qPCR; Sequencing; Sanger sequencing; Hybridization
Introduction
Since late 2019, the COVID-19 crisis has led to a dramatic loss of human lives and presents an unprecedented challenge to public health and socio-economic status worldwide. According to Real Time World Statistics, as of May 16, 2021, there were >163 million cumulative human cases with over 3.3 million deaths due to COVID-19 since the start of the crisis [1]. It has been recognized from the very beginning of the crisis that accurate diagnosis as well as identification and management of symptomatic and asymptomatic COVID-19 cases are important for proper patient care and for making appropriate public health policies. In general, commercial tests based on reverse transcription-quantitative polymerase chain reaction, or RT-qPCR, have been used for laboratory diagnosis. This technology is often referred to as the “gold standard” diagnostic method and mislabeled as “RT-PCR” [2]. However, RT-qPCR tests for SARS-CoV-2, the causative agent of COVID-19, have been found to generate both false-positive and false-negative test results [3–5].
False-negative RT-qPCR test results were more apparent among a large number of seriously ill COVID-19 patients admitted to hospitals. For example, in the month from January 6 to February 6, 2020, 308 of 413 (75%) patients admitted to a Wuhan teaching hospital in China who tested negative for SARS-CoV-2 by an RT-qPCR assay were found to have chest CT findings considered diagnostic of COVID-19 pneumonia [6]. According to Bayes’ theorem, the positive or negative predictive value of an imperfect test depends on the prevalence of the condition in the population being tested and the sensitivity of the test. Among a patient population with a 50% pretest probability of COVID-19 infection, the post-test probability of infection with a negative test result based on using a laboratory test with 70% sensitivity would be 23% [7]. During the first wave of the outbreak, the pretest probability of COVID-19 infection among the serious patients referred to the major teaching hospital in Wuhan might well have exceeded 50% while these patients were being tested by an RT-qPCR with sensitivity below 70%. It is well known that the sensitivity of RT-qPCR assays varies widely, and may drop to 0–50% when the target RNA is lowered to 5–50 copies per reaction [8]. A combination of high COVID-19 prevalence among admitted patients and low RT-qPCR test sensitivity is bound to generate a large number of false-negative test results in a hospital setting.
While false-negative results of an imperfect test were the major concern at the hospitals admitting severe COVID-19 patients, false-positive test results became more evident if the imperfect tests were used to screen a population with low COVID-19 prevalence. For example, when two different nucleic acid assays were used to re-test 52 first-time SARS-CoV-2-positive samples selected from a pool of tests in a general population with low infection prevalence, SARS-CoV-2 RNA was found in only 29 samples on the second test, suggesting true SARS-CoV-2 infection in only 56% of these positive cases [9]. However, it is hard to establish whether the initial positive tests, or the negative results after retesting were the true outcome. Minimization of false-positive SARS-CoV-2 RNA test results is imperative for reducing unnecessary anxiety among the population, and to prevent shutting down schools and workplaces unnecessarily as businesses try to resume normal operations in the community.
Mass screening of the general population for SARS-CoV-2 infection with an imperfect RT-qPCR test may have undesirable impacts on people’s lives. For one notable example, in 2020 seventy-seven (77) football players in the USA were found to be positive for SARS-CoV-2 and barred from playing. However, re-testing confirmed that the results of the first tests were all false positives [10]. Similar cases have been reported in Europe involving professional soccer players.
The US Food and Drug Administration (FDA) has officially alerted clinical laboratory staff and health care providers of an increased risk of false-positive results with some of these commercial test kits granted Emergency Use Authorization (EUA) [11, 12].
After the FDA issued an alert that the Roche Molecular Systems, Inc. Cobas SARS-CoV-2 test kits may generate false positive results, Roche admitted that abnormal PCR cycling in the reaction tubes may produce erroneous results. It is thought to only occur sporadically and may be caused by multiple factors happening at the same time, such as hardware positioning, volume movement, abnormal PCR amplification curves and curve interpretation [13].
On December 14, 2020 the WHO issued an official document, which stated “WHO has received user feedback on an elevated risk for false SARS-CoV-2 results when testing specimens using RT-PCR reagents on open systems.” The WHO further stated “In some circumstances, the distinction between background noise and actual presence of the target virus is difficult to ascertain” and advised that “the cut-off should be manually adjusted to ensure that specimens with high Ct values are not incorrectly assigned SARS-CoV-2 detected due to background noise.” [14]
A subsequent document titled “Genomic sequencing of SARS-CoV-2: a guide to implementation for maximum impact on public health. Geneva: World Health Organization”, published on January 8th, 2021, further advised that “SARS-CoV-2 gene sequencing can be used in many different areas, including improved diagnostics, development of countermeasures, and investigation of disease epidemiology.” [15]
In view of the concerns raised by the social media, the FDA and the WHO about the reliabilities of the RT-qPCR tests currently being used for the presumptive detection of SARS-CoV-2 in clinical specimens, it is expected that the public asks for clearance with respect to the science behind the RT-qPCR technology and whether it is suitable as a tool for diagnostics of SARS-CoV-2 infections. Open evidence-based scientific discussion without censorship should be encouraged to address these questions.
However, in a recent Editorial Commentary by Bustin et al [16] the voices criticizing the RT-qPCR assay as a suitable tool for the molecular diagnosis of SARS-CoV-2 infections were dismissed as “misinformation, outright lies, tendentious reporting and circulation of malicious alternative facts” without supportive data for such accusations. Using the commentary as reference, Bustin was quoted in a Diagnostics Network interview as saying that “PCR tests can distinguish the original virus from the variant even if they differ by only a single nucleotide change (e.g., variant B.1.1.7 N501Y, which has a nucleotide change of A to T within the sequence coding for the spike protein) [17]”. Nevertheless, Bustin did not present any scientific evidence for these claims in his commentary [16], neither in his interview [17].
Bustin’s unscientific prerogatives should not stand unchallenged. To balance his biased Editorial Commentary [16] and to inform people who are not well-educated in molecular sciences, this paper presents experimental data and evidence-based arguments to support the WHO’s position and those who have raised concerns about relying on RT-qPCR assays for the definitive detection of SARS-CoV-2 [18]. It is well known that a distinction between background noise and actual presence of the target virus is sometimes very difficult to ascertain by the RT-qPCR assay alone [14]. Routine partial SARS-CoV-2 gene sequencing should be used to improve diagnostic accuracy, as the WHO advised [15].
Materials and Methods
Materials
The residues of 30 nasopharyngeal swab samples, each in 1–2 mL of VTM, certified to be positive for SARS-CoV-2 N gene RNA by RT-qPCR methods, were purchased from Boca Biolistics Reference Laboratory, Pompano Beach, FL, a commercial reference laboratory endorsed by the FDA as a supplier of clinical samples positive for SARS-CoV-2 by RT-qPCR assays granted Emergency Use Authorization. These samples were collected in the month of October, 2020 from patients with respiratory infection in southern Florida, U.S.A. and were tested twice by two independent CLIA-certified laboratories, initially tested at the time of sample collection from the patients and then re-tested by Boca Biolistics Reference Laboratory before the residues of these positive samples were sold. According to Boca Biolistics Reference Laboratory, all residual samples were stored at -80°C and re-tested with the PerkinElmer® New Coronavirus Nucleic Acid Detection Kit, a real-time RT-PCR in vitro diagnostic test intended for the presumptive qualitative detection of nucleic acid from the SARS-CoV-2 virus. The N gene Ct values for these samples ranged from 14.55 to 36.71. The samples were shipped to Milford Molecular Diagnostics Laboratory in dry ice over-night delivery. The received samples were stored in a -80°C freezer upon receipt and tested within one week.
Methods
RNA extraction and purification, nested RT-PCR amplification and Sanger sequencing of the cDNA PCR amplicon were performed as previously described [29]. In this protocol, we used nested PCR to amplify the target cDNA to increase the detection sensitivity and to prepare very clean templates for DNA sequencing. To perform nested PCR, a very small portion of the first (primary) PCR products is transferred to the second (nested) PCR for a two-round PCR amplification. Each round of PCR is limited to 30 cycles because after 30 heating cycles the partially degraded DNA polymerase may generate more errors, making unwanted PCR products when there are nontarget DNAs and RNAs in the PCR matrix. In the second round (nested) PCR, the ratio of target DNA/non-target DNA is markedly increased with new DNA polymerase, creating a condition favoring highly effective amplification of the target DNA. As a result, we always rely on the nested PCR for detection and for preparation of the template for DNA sequencing. In other words, we routinely run a total of 60 cycles for nested PCR amplifiation. In contrast, RT-qPCR is never allowed to run 60 cycles
Results and Discussion
qPCR is not PCR
All nucleic acid tests involve determination of the order of the four nucleotides in a target segment of genetic DNA or RNA. PCR was invented to make templates in vitro for DNA sequencing. “The polymerase chain reaction has made genetic sequences much more easily available”, a statement made by Appenzeller in 1990 [19], three years before Kary Mullis, the inventor of PCR, was awarded a Nobel Prize. Prior to the development of PCR, scientists had to clone DNAs into E. coli to generate a mass of identical DNA molecules for nucleotide sequence analysis. The PCR method can be defined as a chemical process of primer-defined, template-directed, exponential enzymatic polymerization of nucleotides to replicate target DNA in a test tube. In molecular diagnostics, PCR is used to specifically amplify the target DNA present in a complex clinical sample, preferably with minimal errors during enzymatic incorporation of dNTPs to the 3’ end of a growing ssDNA to ensure that the dNTPs are correctly incorporated as directed by the template. Therefore, a high-fidelity DNA polymerase is needed to serve this purpose. A consensus PCR primer harboring a few mismatched nucleotides is tolerable and sometimes preferred for a broad-spectrum coverage of target sequence variants [20], as long as unintended, nonspecific PCR amplicons are eliminated at the confirmatory stage by DNA sequencing.
Diagnostic qPCR and RT-qPCR exploit the PCR process as a tool to measure the rate of cleavage and degradation of a single-stranded DNA (ssDNA) “probe”, which binds to the target DNA and to the PCR copies of that target DNA. The degradation of the probe is caused by the 5’-3’ exonuclease activity of the Taq DNA polymerase during primer extension in the PCR process. Commonly, a hydrolysis probe is used, which incorporates a fluorophore attached to the 5’ end and a quencher attached to the 3’ end of the probe (Most TaqMan® probes are based on this principle, including the one used in the SARS-CoV-2 RT-qPCR test kits.). Fluorescence Resonance Energy Transfer (FRET) prevents fluorescence emission of the fluorophore due to proximity of the quencher while the probe is intact. The degradation of the probe by the Taq polymerase separates the fluorophore from the quencher, thus allowing fluorescence of the fluorophore. This leads to a PCR amplification-dependent increase in fluorescence and can be used as a measure for the amount of amplified PCR product. In other words, diagnostic qPCR uses the PCR process to test if a DNA/DNA binding (hybridization) has taken place between two ssDNA molecules (One of them is the fluorescent dye-labeled probe, the other the target DNA or PCR copies of the target DNA). For a qPCR to work properly as a diagnostic tool, the primers and the probe should all bind to their respective sequences present in the target ssDNA and with fully matching base pairs, so that a fluorescence signal will be emitted only after PCR amplification-dependent degradation of the probe.
However, when complex clinical specimens are tested for diagnostic purposes, DNA/DNA hybridization does not always involve two fully matched sequences. This has been demonstrated in DNA/DNA hybridization studies, in which a set of binding partners (6 nucleotides (nt) to 21 nt in length) to a 50-mer oligonucleotide were probed. It was observed that stable hybridization DNA/DNA duplexes were formed when only 12 of 50 bp matched. This resulted in the appearance of significant signals from an unintended binding partner, in the absence of the intended fully matched DNA target [21].
During PCR amplification, each newly synthesized copy of ssDNA is identical to or the complementary mirror image of the template dsDNA, which is flanked by the two PCR primers. The occurrence of Single Nucleotide Polymorphisms (SNPs) or insertion/deletion (indel) mutations in the target DNA between the two PCR primers does not affect the PCR/DNA sequencing-based nucleic acid diagnostics. Such variants (or mutants) will be detected and confirmed at the stage of DNA sequencing. The only goal of PCR is to produce an error-free molecular mass of DNA for sequencing analysis. The computer-generated DNA sequence is submitted to the GenBank electronically for Basic Local Alignment Search Tool (BLAST) analysis. The BLAST report returned from GenBank identifies the nature of the genetic sequence detected by comparing the submitted sequence with the DNA sequences previously deposited in the GenBank databases. Used this way, there is no human interpretative bias involved in molecular detection of SARS-CoV-2 with the PCR amplification followed by Sanger sequencing technology (i.e. when its RNA genome was reverse transcribed into cDNA prior to PCR amplification).
To obtain an accurate test result with the qPCR or RT-qPCR method, the probe must be carefully designed by knowledgeable bio-scientists. It is standard that the nucleotide sequence of the ssDNA probe is the perfect mirror image of the target DNA with 100% base-pair complementarity. Further, the actual binding between the ssDNA probe and the target DNA must take place before the PCR amplification-dependent breakdown of the probe occurs to emit the fluorescence signal that is recorded by the fluorometer. As stated in the Commentary by Bustin et al [16], “These are the SARS-CoV-2-specific primers and probe, which must be 100% specific for the virus and so amplify only viral sequences”. However, these authors failed to point out that even in the widely promoted RT-qPCR protocol cited in their references [22] primers and probes are not 100% specific for the target SARS-CoV-2 gene sequence.
RNA viruses, such as SARS-CoV-2, HIV and influenza, tend to pick up mutations quickly as they are copied inside their hosts because the enzymes that copy RNA are prone to making errors. As of May 14, 2021, the annotated numbers of SNP mutation types reached 28,397 worldwide [23]. As the full genome of SARS-CoV-2 Wuhan-Hu-1 has 29,863 nucleotides (GenBank Sequence ID: NC_045512.2), this number means that over 95% of the 29,863 nucleotides in the genome of the Wuhan-Hu-1 have been found to have mutated at least once by DNA sequencing since the COVID-19 outbreak. Inserting the sequences of 2 of the 3 probes, namely the 27-base Nsp 10 probe and the 26-base N-gene probe, which were designed by Bustin et al [16], into the GenBank for a cursory search revealed that single nucleotide mutation has taken place in 21 of the 27 nucleotides covered by the Nsp 10 probe and in all of the 26 nucleotides covered by the N-gene probe. Mutation in 1 of the 26 nucleotides targeted by the N-gene probe was found in 138,853 of the 521,581 SARS-CoV-2 isolates, which had been sequenced and annotated worldwide. Samples of these mutations were retrieved and cropped from the GenBank DNA sequence database, and are presented in figure 1.
Each probe-targeted sequence is flanked by a pair of PCR primer-binding sequences in boldface and gray-shaded. The two wildtype sequences targeted by the probes were copied from GenBank sequence ID: NC_45512.2, and pasted as reference. The changed nucleotide within the probe sequence in the mutants is in red color. The nucleotide “n” at position 13207 in SARS-CoV-2 Sequence ID: MW705415.1 means that the base “c” originally annotated for the Wuhan-HU-1 strain at this position has been found to mutate into base “a”, “g” or “t”.
In molecular sciences, it is common knowledge that success of DNA hybridization is dependent on the length, composition and sequence of the probe, as well as the composition of the hybridization buffer, temperature of hybridization reactions and concentrations of both target strand and probe. In order to use one single probe to detect all strains of SARS-CoV-2 with nucleotide mutations in the target sequence, the buffer and temperature in the PCR mixture must be less stringent, so that the probe can bind to the mutated sequences. However, with less stringent conditions, Bustin’s N-gene probe may also bind the N gene of bat coronavirus RaTG13 (Sequence ID: MN996532.2; figure 1), which has one base mismatch in this 26-base target sequence compared with some SARS-CoV-2 mutants (Figure 1). As shown in figure 1, the sequences of the two PCR primer-binding sites between bat coronavirus RaTG13 and those of the SARS-CoV-2 strains listed in Figure 1 are identical. It is obvious that the RT-qPCR assays based on the N-gene primers and the probe described by Bustin et al cannot differentiate bat coronavirus RaTG13 from SARS-CoV-2. DNA sequencing of the PCR amplicon is the only way to obtain a conclusive result.
Further, the Nsp-10 probe and the N-gene probe also harbor a 15-base sequence and a 16-base sequence, respectively, that are identical to some segments of human genomic DNA (Sequence ID: NM_001324512.2 and Sequence ID: NM_173484.3). Since it takes only 12 bp of complementary sequence with an unintended binding partner to form a stable partial duplex [21], under the right conditions the Nsp-10 and N-gene probes bound to human genomic DNA in clinical samples are capable of undergoing PCR amplification-dependent breakdown when this segment of human genomic DNA should become an unintended PCR template. A combination of these unintended events by chance may lead to emission of unwanted fluorescence signals, causing confusing or false-positive RT-qPCR test results.

Figure 1: DNA sequences of the Nsp-10 probe and N-gene probe regions and their respective flanking PCR primer pair described by Bustin et al in their RT-qPCR protocol [16] and several sampled sequences retrieved from the GenBank database. The 27 bases and the 26 bases targeted by the Nsp-10 probe and the N-gene probe, respectively, are highlighted yellow.
The sequence of the forward primer designed by Bustin et al for N-gene amplification is GCTGCTAGACAGATTGAAC [16]. The authors probably intended to use this PCR primer to amplify the sequence of certain SARS-CoV-2 mutants, for example, the GenBank Sequence ID: MT308984.1, as shown in Figure 2, because none of the SARS-CoV-2 N gene sequences presented in Figure 1, including that from the Wuhan-Hu-1 reference Sequence ID. NC_045512.2, fully match this forward primer sequence. The nucleotide at reference position 28945 is a constant “t” base (in red) on all SARS-CoV-2 strains that have been sequenced and annotated in the GenBank database. Submission of this 19-base forward PCR primer sequence to the GenBank for BLAST analysis elicited a report indicating that the DNA sequence of this primer matches fully with a segment of genomic nucleic acids in Calothrix, a genus of cyanobacteria generally found in freshwater but also found in human respiratory tract [24]. The same sequence also fully matches the sequence of the N-gene in several bat SARS-like coronavirus isolates as shown in figure 2.

Figure 2: Nonspecific design of the SARS-CoV-2 N-gene RT-qPCR Forward Primer may also yield positive results for Cyanobacteria and other Coronavirus strains.
Using primers with mismatched nucleotides to perform RT-qPCR may have serious impacts on the fluorescence growth curve and the Ct values. Therefore, even the primers and probes designed by RT-qPCR specialists may lead to generation of unintended PCR products, such as an unintended PCR amplicon of a genetic sequence from Cyanobacteria that may be found in human respiratory tract.
Real-time or quantitative PCR (qPCR) was first described in 1993 to monitor the accumulation of double-stranded DNA (dsDNA) being generated in each PCR, using the increase in the fluorescence of ethidium bromide (EtBr) that results from its binding to dsDNA as the PCR products [25]. The kinetics of fluorescence accumulation during thermocycling is directly related to the starting number of DNA copies in the PCR mixture. The basic principle dictates that the fewer cycles necessary to produce a detectable fluorescence, the greater the number of target sequences in the original sample being tested. Results obtained with this approach indicate that a kinetic approach to PCR analysis can quantitate the copy numbers of a known dsDNA in the mixture when there are no other interfering DNAs in the PCR mixture [25]. This process is referred to as dye-based qPCR for quantitation of small amounts of target DNA known to exist in a sample. It is widely acknowledged as the most sensitive method to quantify minute amounts of nucleic acids of known DNA sequence and its applications split into two main types referred to as: relative and absolute quantification. In relative quantification the analyte, often reverse-transcribed mRNA or microRNA, is quantified relative to an endogenous reference. In absolute quantification the targeted nucleic acid (the analyte) is measured relative to a set of standards used to construct a standard curve [26]. It should be emphasized that qPCR was not designed to determine whether a target DNA is present or absent in the sample being tested, neither was it designed to detect a single target DNA molecule, which seems to be the goal in molecular diagnosis of infectious diseases.
Later, the dye-based qPCR was converted to a probe-based qPCR, which paved the way for diagnostic assays to detect infectious disease agent nucleic acid in an absolute way (“yes/no” or “positive/negative”, meaning “infected/not infected”). In these assays, instead of a free dye, such as EtBr, a target-specific ssDNA probe of about 25 bases and complementary to the target DNA sequence is used. This method is, however, not as reliable as dye-based qPCR. The flaws of the probe-based qPCR for molecular diagnosis of infectious diseases are being exposed when this platform is used for detection of SARS-CoV-2 during the COVID-19 pandemic.
In the recent Commentary, Bustin et al write: “RT-qPCR has revolutionized the diagnosis of infectious diseases [16].” Further they write: “The sensitivity and specificity of the technique has allowed it to identify active or recent infection by detecting the genomes of disease-associated pathogens, including viruses” [16]. Although the authors cite a review article on qPCR [27], it is unclear how this paper supports their claims. Careful reading of the cited reference [27] demonstrates that it describes two kinds of quantitative PCR methods: 1) relative quantitation, and 2) absolute quantitation. Relative quantitation describes changes in the amount of a target sequence compared with its level in a relative matrix, also referred to as endogenous reference. Absolute quantitation states the exact copy number of a nucleic acid target present in the sample in relation to a specific unit on an absolute standard curve. Scientifically speaking, a “yes/no” assay to detect viruses is an absolute quantitative PCR, because there is no endogenous reference in the matrix to compare with. But in chemical quantitative analysis, the spacing between the zero calibrator and the Lower Limit of Quantitation (LLOQ) of an analyte is extremely difficult to determine [28]. In other words, the technology depending on RT-qPCR for the definitive detection of one single copy of SARS-CoV-2 target nucleic acid in an RT-qPCR mixture is not reliable. This is particularly true when the viral load is low. That is why the WHO stated in their official release: “In some circumstances, the distinction between background noise and actual presence of the target virus is difficult to ascertain” [14].
It cannot be emphasized enough that scientifically speaking, PCR, i.e. polymerase chain reaction, is a technology that exploits the function of an enzyme (DNA polymerase), which can exponentially and accurately duplicate target DNA thus making it available for nucleotide sequence analysis [19]. The purpose of PCR is to generate a mass of identical ds DNA for analysis.
In contrast, although qPCR and RT-qPCR use “PCR” as a part to create a new compound word, qPCR and RT-qPCR are de facto probe-based hybridization technologies. They measure the rate of hydrolysis of a single-stranded DNA probe with a known nucleotide sequence, whereas the probe is attached (=hybridized) to another single-stranded DNA that serves as the template in the enzymatic primer extension reaction. In qPCR, the rate of probe hydrolysis is measured and expressed as “Ct value”. Ct values do not determine or confirm genetic nucleic acid sequence of the PCR template. In fact, the DNA sequences of the PCR products generated by qPCR, especially those generated during the late cycles of the amplification process may not be identical to those of the initial template. This is due to the DNA polymerase used in the PCR reaction (Taq), which deteriorates over time and its copying fidelity is not monitored by DNA sequencing. Therefore, qPCR is not PCR although qPCR uses the PCR technology to measure the rate of probe degradation. In a similar way, a straightjacket is not a jacket even though both are garments put on a human body. This distinction is crucial in reading publications about accuracy of molecular detection of SARS-CoV-2. The major differences between qPCR and PCR are summarized as follows.
A specific probe-based RT-qPCR assay (for instance for SARS-CoV-2) requires a unique segment of gene sequence with three conserved sites, two for the PCR primers and one for the probe. Identification of three conserved regions within a short region of the virus genome, typically <100 bases, may not always be possible in view of the frequency of single nucleotide mutations in this RNA virus (see Figure 1). As stated above, >95% of the nucleotides in the SARS-CoV-2 Wuhan-Hu-1 genome have been shown to mutate at least once.
The diagnostic technology based on PCR amplification followed by Sanger sequencing [29] needs only two 21-base moderately conserved sequences for the primer sites, which flank a 300- to 400- base segment of a unique SARS-CoV-2 gene sequence as the template. Mutations in the target or sequence variations do not affect detection sensitivity or specificity by Sanger sequencing. All interprimer single nucleotide mutations, deletions or insertions can be detected by a bi-directional sequencing of the PCR amplicon. Any PCR false-positive results are eliminated at the stage of Sanger sequencing.
Unintended PCR products in SARS-CoV-2 assay
In order to investigate whether RT-qPCR generates false positives, 30 nasopharyngeal swab samples, which were certified to be positive for SARS-CoV-2 N gene (see Materials Section), were subjected to Sanger sequencing. These samples were residual patient samples sold by a company endorsed by the FDA [30] and the FDA also recommends that Sanger sequencing may be used to investigate false results generated by RT-qPCR assays under emergency use authorization [31].
The RNA extracts of the 30 samples were prepared and a 1-µL aliquot of each RNA extract was amplified by a pair of nested PCR primers to generate a 398-bp cDNA amplicon of the SARS-CoV-2 N gene. The 398-bp cDNA amplicon was then used as the template for automated Sanger sequencing, using a method previously described [29]. The sequences of the forward and reverse nested PCR primers are 5’-CAATCCTGCTAACAATGCTGC-3’ and 5’-TTTGTTCTGGACCACGTCTGC-3’, respectively.
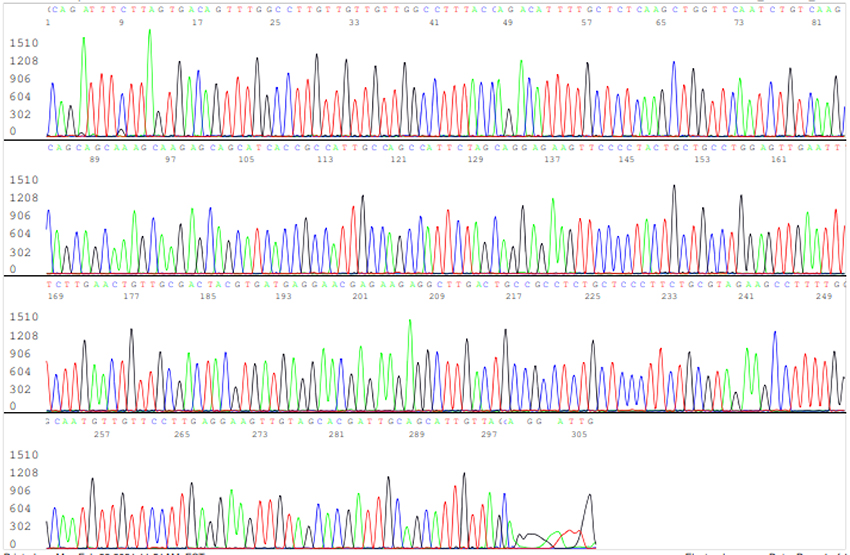
The results of the nested RT-PCR amplification followed by Sanger sequencing assay showed that only 16 out of the 30 RT-qPCR-positive samples were true positives (53%). A sample of the true-positive computer-generated base-calling sequence electropherogram is illustrated in Figure 3.
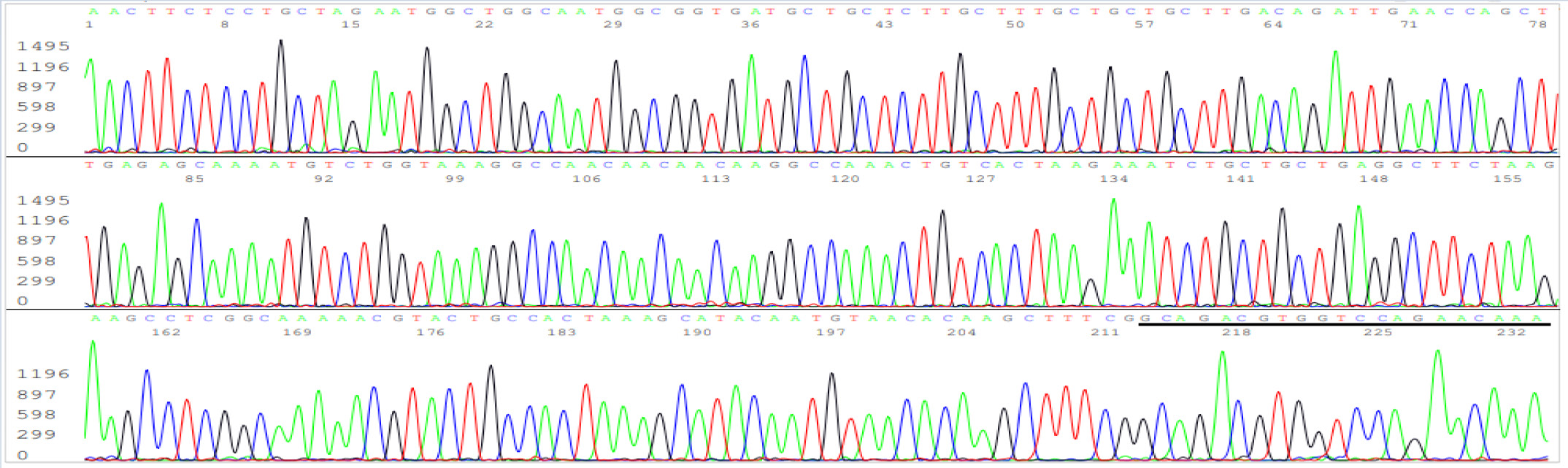
Figure 3: Electropherogram showing definitive detection of SARS-CoV-2 N gene in a true-positive sample. The figure represents one of the two independent bi-directional sequencing electropherograms of a 398-bp N gene cDNA PCR amplicon. This sequence was generated with a forward nested PCR primer. The reverse primer-binding site is underlined (the complementary sequence of the reverse primer is GCAGACGTGGTCCAGAACAAA).
For the other 14 samples on which Sanger sequencing did not generate a DNA sequence, some of their PCR products were visualized as smears or as poorly defined bands at agarose gel electrophoresis. Sanger sequencing showed that these PCR products were composed of randomly terminated DNA fragments, mostly of unidentifiable source (Figure 4). Nevertheless, further analyses showed that some of the amplified PCR products could be traced back to chimeric DNA (figure 5) and human genomic DNA (figure 6).
Collectively, these data demonstrate that in the presence of SARS-CoV-2 genomic RNA during RT-PCR, the PCR primers amplified the intended 398-base target N gene segment to be used as the template for Sanger sequencing (see Figure 3). However, in the absence of a fully matched preferred template, PCR primers can attach to partially matched ssDNA sequences to initiate an enzymatic primer extension. An unintended PCR amplicon (PCR product) will be formed if the second PCR primer also anneals to the complementary ssDNA from the opposite end of an unintended template to initiate a primer extension (see Figures 4 and 5). Identification of the binding site sequence for the reverse PCR primer on an electropherogram indicates annealing of the forward PCR primer at the opposite end of an unintended template. The GenBank BLAST report confirmed that one of the amplified DNA segments was caused by a gene present on human chromosome 1 (figure 6). Further, 10 common bases (figure 6, in red) were observed between a 21-base segment of this human gene and the 21-base primer (yellow-highlighted) designed for SARS-CoV-2 N gene amplification. Notably, six of the 10 commonly shared nucleotides are located in the 3’ end of the primer. The result is a chimeric DNA composed of a virus genomic sequence and a human genomic sequence through joining of the 6-bases that both have in common.
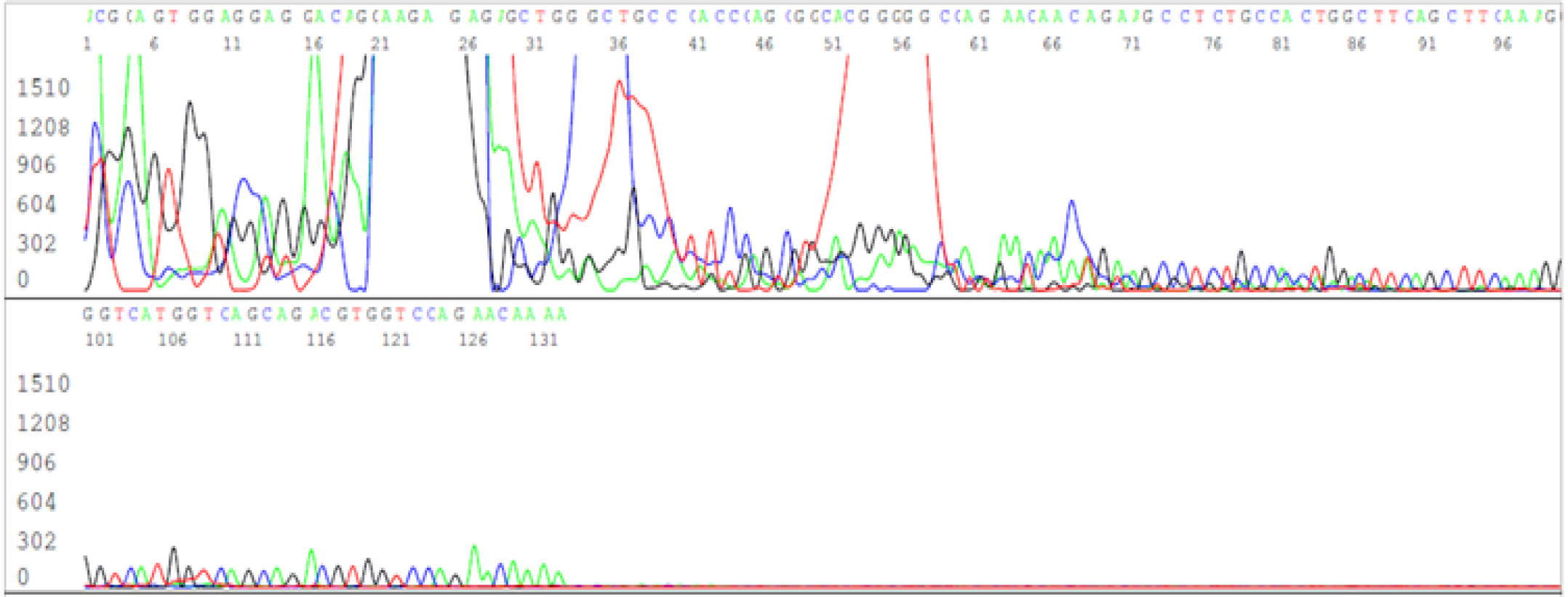
Figure 4: Electropherogram showing numerous unintended PCR products on a negative sample of unknown source.
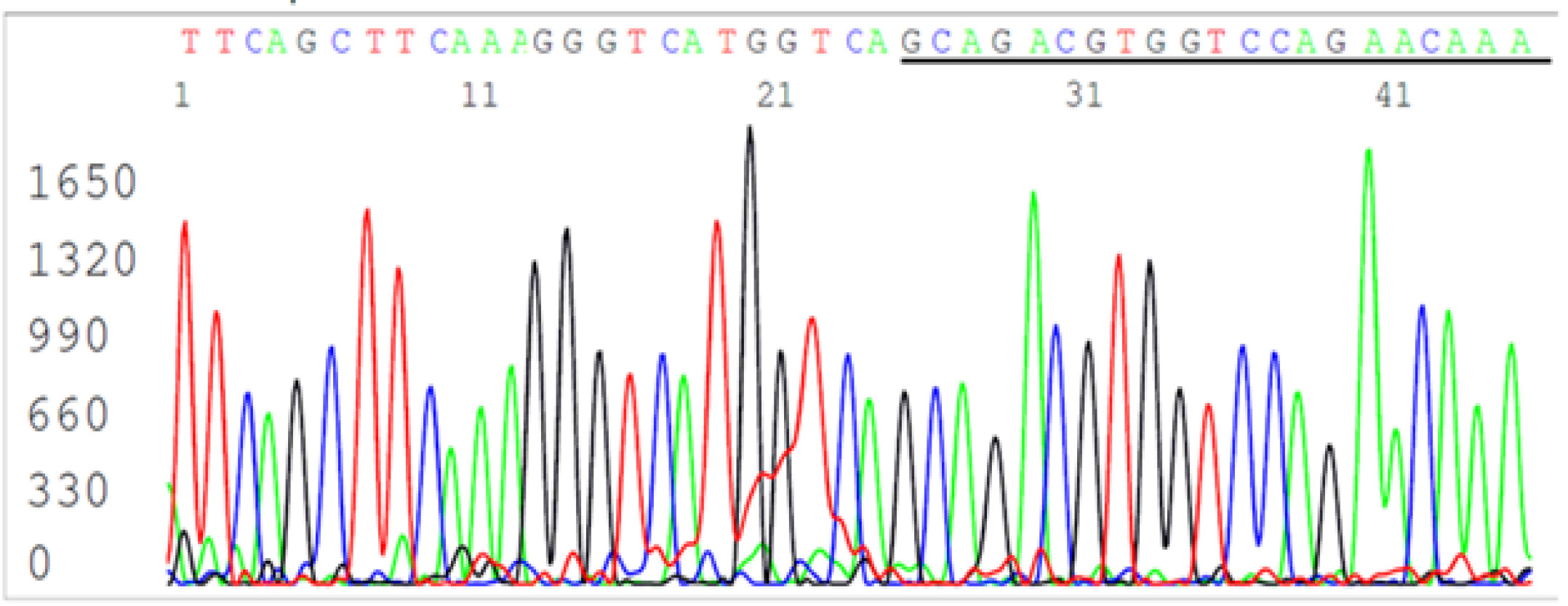
Figure 5: Magnification of last 45 bases of the computer-readable sequence in Figure 4 showing a chimeric DNA sequence (the complementary sequence of the SARS-CoV-2 N gene reverse primer is underlined).
Figure 6: BLAST report from the GenBank showing that the DNA sequence downstream of the SARS-CoV-2 N gene reverse PCR primer is a segment of human genomic DNA.
A nasopharyngeal swab contains a variety of nucleic acids, not only derived from human cells, but also from a plethora of viruses, bacteria and fungi normally residing in the human respiratory tract. The nature and quantity of these nucleic acids in the respiratory tract vary from person to person. Therfore, when PCR is used as a tool to amplify a target DNA segment in such a complex clinical specimen, the results must be taken with caution.
Likewise, when RT-qPCR is used for diagnostics, it may generate cumulative irrelevant fluorescence signals leading to false-positive results even when there is no target genetic material in a patient’s sample. The function of DNA polymerases is to add nucleotides to the 3’ end of an oligonucleotide or a polynucleotide chain of a partial duplex. As reported more than 20 years ago, it takes only 6 bp of complementary sequence at the 3’ end of a primer to initiate an enzymatic primer extension [32]. This observation has been confirmed previously by studies of non-B conformational changes when HPV L1 DNA is bound to an aluminum salt in vaccine formulation [33]. The results presented in Figures 4–6 support these earlier findings.
It is also reported that nucleotide mismatches at the 3’ terminus of a primer do not always prevent enzymatic primer extensions [34, 35]. One of the important factors to yield PCR products is the relative efficiency with which the DNA polymerase extends from a mismatched primer-template duplex. It is important to realize that once extension from a mismatched primer occurs, the resultant product and the complement synthesized in subsequent PCR cycles are fully matched with both primers and will produce unintended products [34].
Amplification of non-specific products in RT-qPCR assays occurs frequently and is unrelated to Ct or PCR efficiency values. The frequency of the amplification of the correct product and the artifact, as well as the valid quantification of the correct product, depends on the concentration of the non-template DNAs [36], which are unpredictable in respiratory tract specimens.
In summary, although PCR plays a very important role in nucleic acid-based diagnostics, it is compulsary to analyze PCR products by DNA sequencing if accurate molecular diagnosis of infectious diseases is desired. The sequencing electropherograms of these 30 RT-qPCR positive samples are presented as a Supplemental File to support this statement.
Flaws of Diagnostic qPCR exposed in COVID-19 pandemic
RT-qPCR assay has its inherent flaws. As discussed above, even probes and PCR primers designed by RT-qPCR specialists for SARS-CoV-2 detection can be flawed, in particular when highly sensitive and specific sequence-based diagnostics is needed [16]. To overcome the low sensitivity due to mismatched bases in the probe or primers (Figure 1), the RT-qPCR requires 40 cycles of amplification [16]. Nevertheless, in writing guidelines for the industry on quantitative real-time PCR, the same group of qPCR specialists defined that “…, specificity must be validated empirically with direct experimental evidence (electrophoresis gel, melting profile, DNA sequencing, amplicon size, and/or restriction enzyme digestion)” and Ct “values >40 are suspect because of the implied low efficiency and generally should not be reported” [37]. This discrepancy is highly peculiar and should be addressed by these specialists in an open discussion. It should also be emphasized that the RT-qPCR test kits, which were developed by these specialists or granted emergency use authorization by the FDA, have not been validated. So, we do not know whether the qPCR products generated by these test kits are specifically and solely SARS-CoV-2 genomic sequences.
Further, Bustin et al argue: “RT-qPCR unquestionably provides the most reliable, rapid, sensitive, specific and flexible means of detecting SARS-CoV-2 RNA” and “RT-qPCR has revolutionized the diagnosis of infectious diseases. The sensitivity and specificity of the technique has allowed it to identify active or recent infection by detecting the genomes of disease-associated pathogens…” [16]. Here, it is important to note that such an over-confidence in the reliability of the RT-qPCR method for SARS-CoV-2 diagnostics has misled and will further mislead health care policy makers to impose grave socio-economic measures upon our society, which may have severe but unforeseen repercussions. Therefore, PCR scientists should be more cautious in their reports to the mainstream media. In particular, unscientific statements, such as “PCR tests [can be used] to distinguish single nucleotide change in spike protein gene, such as that occurring in N501Y with A>T mutation” [17] should be avoided.
The FDA has published clear guidance on molecular diagnostics for RNA virus infections. In a document titled “Nucleic Acid Amplification Assay for the Detection of Enterovirus RNA - Class II Special Controls Guidance for Industry and FDA Staff”, the FDA advises “Detection of an EV genome in CSF by two different well-characterized and validated nucleic acid amplification tests (NAAT). The NAAT primers pairs should generate amplicons from different genomic regions. One of the NAAT assays should provide sequence information. Bi-directional sequencing should be performed on both strands of the amplicon and the generated sequence should be of an acceptable quality (quality score of 40 or higher as measured by PHRED or similar software packages) and should match the reference or consensus sequence.” [38]
Further, for the detection of the human papillomavirus (HPV), the FDA also advises to use a conventional PCR detection followed by Sanger sequencing on both strands of the PCR amplicon (bi-directional sequencing), which should at least contain 100 contiguous bases. When these conditions are met the method is acceptable as a valid diagnostic for HPV infection, provided that the sequence matches the reference or consensus sequence, e.g. with an Expected Value (E-Value) <10–30 for the specific HPV DNA target based on a BLAST search of the GenBank database [39].
None of the commercial SARS-CoV-2 RT-qPCR test kits granted EUA meet these requirements stipulated in the FDA guidance for Nucleic Acid Amplification Assay for the Detection enterovirus or HPV infections. In view of the recently reported increased mortality hazard ratio associated with infection by certain emerging variants of SARS-CoV-2 with S gene mutations [40], routine PCR amplification followed by bi-directional sequencing of the spike protein gene in part or in full length is valuable not only for proper patient management, but also for making appropriate public health policies. With the current population-wide testing scheme it is not practical to routinely perform full-length genome sequencing on every positive specimen. Nevertheless, routine sequencing of a short segment of the S gene covering the ACE2 receptor binding domain for possible key single nucleotide mutations on every positive sample is feasible. An example of bi-directional sequencing of a segment of the S gene, including the L452R, S477N, E484K and N501Y mutation sites, is illustrated in Figures 7 and 8.
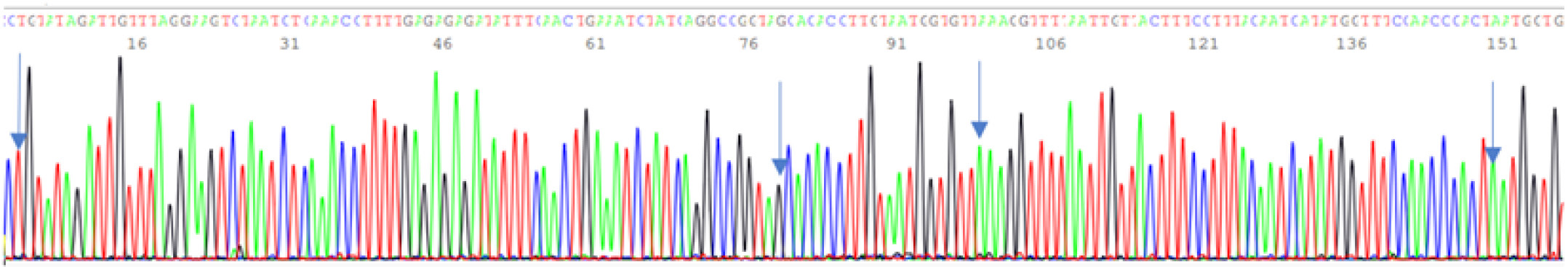
Figure 7: Forward Sanger sequencing electropherogram showing negative L452R (leucine to arginine), negative S477N (serine to asparagine), positive E484K (glutamate to lysine) and negative N501Y (asparagine to tyrosine). The four arrows from left to right are pointing to a wildtype base T at position 22917, a wildtype base G at position 22992, a G>A mutation(E484K) at position 23012 and a wildtype base A at position 23063.
Currently, the socalled “UK variant” of the SARS-CoV-2 virus (named B.1.1.7) is studied using the TaqPath RT-qPCR assay as a proxy measure of VOC-202012/1 infection. It is based on positive detection of both the N gene (Ct <30) and the ORF1ab gene (Ct <30), and negative detection of the S gene in a clinical sample [40], which is a presumptive molecular diagnosis of a VOC-202012/1 infection. Intriguingly, the test kit manufacturer recommends to confirm detection of the “UK variant” using Sanger sequencing of the S gene to detect the 69–70del [41]. This shows that also in the industry Sanger sequencing is considered the method of choice for definitive detection of emerging variants. The claim that RT-qPCR tests can distinguish between the original virus and novel variants, even if they differ by only a single nucleotide change, [16,17] is beyond evidence-based science.
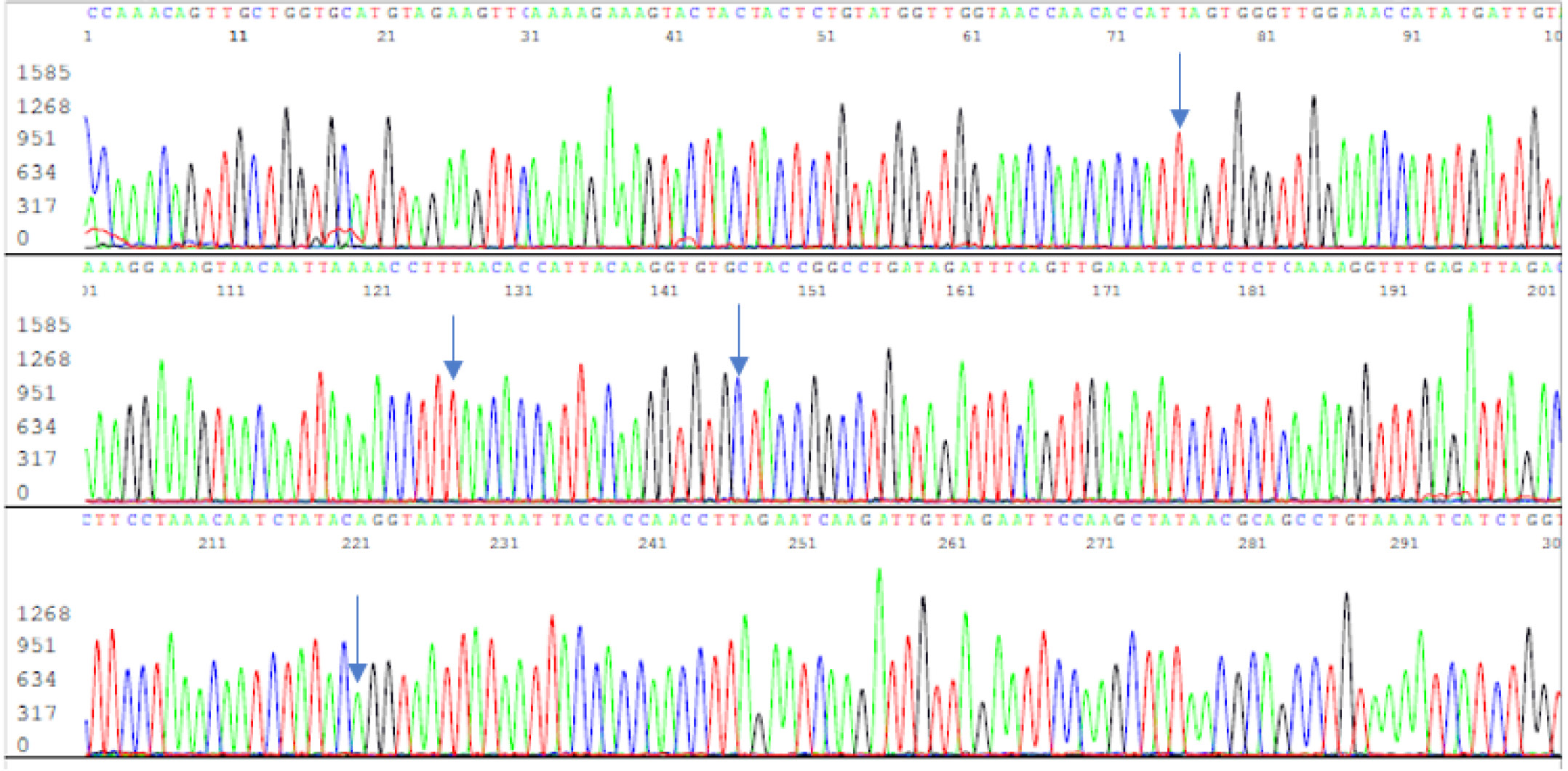
Figure 8: A reverse Sanger sequencing electropherogram of the PCR amplicon used to generate the sequence illustrated in Figure 7. The four arrows are pointing to the reverse complementary bases noted in Figure 7 for validation of a single E484K mutation.
The observation that qPCR lacks diagnostic specificity is well known among those in the field of infectious diseases. For example, after spending years of effort trying to develop a qPCR test for Lyme disease, a tick-borne infectious disease caused by Borrelia burgdorferi, a group of experts concluded that until the specificity of qPCR techniques is determined, the clinical utility of such testing relative to other testing modalities will remain uncertain [42]. A CDC expert, Christina A. Nelson, MD, MPH, emphatically stated on record, while answering a self-posed question: Is PCR useful for the diagnosis of Lyme disease? In general, the answer is no [43].
qPCR has also been used in the Roche Cobas system for the detection of human papillomavirus (HPV) in cervical screening along with Pap smear. Since HPV is a DNA virus, there is no need for reverse transcriptase to detect HPV DNA. All HPV tests are known to have “built-in false positives” by those who promote the test [44]. After a Human Papilloma Virus (HPV) Proficiency Test conducted by the Department of Health (DOH), New York State (NYS), in October 2015, the summary report showed that compared to the consensus standards the false-positive rates in high-risk HPV detection generated by the Roche Cobas system based on its qPCR platform were found to range from 45.8% to 83.3% [45]. Since most cyto-pathologists who are in charge of the HPV testing have little general molecular biology experience and the test kits were FDA-approved, false-positive and false-negative HPV tests usually go unnoticed or are neglected. This may have caused that thousands of healthy women had to undergo colposcopic biopsies unnecessarily based on these built-in false-positive test results [46]. Fortunately, as a result of the COVID-19 pandemic there has been increased awareness of the false positives of the HPV qPCR tests. As stated by one cyto-pathologit, “because of COVID testing, for which the workhorses are the two platforms (Cobas and Panther) that are also used for HPV testing, there’s been increased awareness of false-positives—which run about 0.7 to 0.35 percent (from just carryover issues) with HPV testing—and false-negatives.”[47]
The “built-in false positives” of the qPCR HPV tests have been statistically explained away, arguing that the benefit of false-positive test results outweighs the harm done by relying on true positives, which may lower the positive rates [44]. While the cyto-pathologists started to look into the HPV false-positive tests, they also confirmed the previous observations that the HPV tests, including those based on qPCR assays, also generated false-negative results [47]. A false-negative HPV screening test poses a more serious liability to the practicing pathologists in cervical cancer prevention. Recently, it was widely reported that a case of false-negative HPV screening test led to development of a fatal cervical cancer in Ireland, which resulted in a damage payment of several million dollars [48]. This in mind, we should also be very cautious with SARS-CoV-2 RT-qPCR test results. The exceptionally high death tolls among the nursing home residents associated with false-positive SARS-CoV-2 RT-qPCR test results are hard to gloss over.
Conclusion
As stated in a recent CDC Advisory titled Nucleic Acid Amplification Tests (NAATs), “a Nucleic Acid Amplification Test, or NAAT, is a type of viral diagnostic test for SARS-CoV-2, the virus that causes COVID-19. NAATs detect genetic material (nucleic acids). NAATs for SARS-CoV-2 specifically identify the RNA (ribonucleic acid) sequences that comprise the genetic material of the virus” [49]. The data presented in this article further emphasize that the technologies depending on probe hybridization, in particular RT-qPCR, cannot accurately and specifically identify the RNA sequences that comprise the genetic material of the virus, especially of a virus with frequently emerging sequence variants. The claim that RT-qPCR tests can distinguish between the original virus and novel variants, even if they differ by only a single nucleotide change [16,17], is not evidence-based and beyond scientfic realism. For definitive molecular detection of SARS-CoV-2 in clinical specimens, routine Sanger sequencing of a 300–400 bp RT-PCR cDNA amplicon of a unique sequence of the viral genome is needed to ensure 100% specificity and to avoid false-positive test results. False-positive SARS-CoV-2 test results can easily create unnecessary panic and may result in negative impacts on local economies. Newly emerging SARS-CoV-2 strains with single nucleotide mutations can only be reliably screened and confirmed by DNA sequencing. Since RT-qPCR depends on fully matching sequences between the template and its primers and probe to generate a positive fluoresence signal, relying on RT-qPCR for molecular diagnosis will produce more false-negative test results as new SARS-CoV-2 sequence variants emerge when the sequences of the primers and probe no longer fully match the sequene of the new target template.
Acknowledgment
The manuscript of this article was initially submitted to the International Journal of Molecular Socieces Section “Molecular Diagnostics” and was rejected by Stephen A. Bustin, Section Editor in Chief, without peer reviews.
Institutional Review Board Statement: Not applicable. The materials tested were commercial products with no patient identifiers.
References
- https://www.worldometers.info/coronavirus/
- Dramé M, Tabue Teguo M, Proye E, Hequet F, Hentzien M, Kanagaratnam L, Godaert L (2020) Should RT-PCR be considered a gold standard in the diagnosis of COVID-19? J Med Virol 92: 2312–2313. [View]
- Rahman H, Carter I, Basile K, Donovan L, Kumar S, etc. (2020)Interpret with caution: An evaluation of the commercial AusDiagnostics versus in-house developed assays for the detection of SARS-CoV-2 virus. J Clin Virol.127: 104374. [View]
- Kelly, Meg, Sarah Cahlan and Elyse Samuels. (2020) What went wrong with coronavirus testing in the U.S. Washington Post. March 30, 2020. [View]
- Willman, David. (2020) CDC coronavirus test kits were likely contaminated, federal review confirms. Washington Post. June 20, 2020. [View]
- Ai T, Yang Z, Hou H, Zhan C, Chen C, Lv W, et al. (2020) Correlation of Chest CT and RT-PCR Testing for Coronavirus Disease 2019 (COVID-19) in China: A Report of 1014 Cases, Radiology. 296: E32-E40. [View]
- Woloshin S, Patel N, Kesselheim AS (2020) -CoV-2 Infection False Negative Tests for SARS - Challenges and Implications. N Engl J Med 383: e38. [View]
- Vogels CBF, Brito AF, Wyllie AL, Fauver JR, Ott IM, Kalinich CC, et al. (2020) Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT-qPCR primer-probe sets. Nat Microbiol. 5: 1299–1305. [View]
- Skittrall JP, Wilson M, Smielewska AA, Parmar S, Fortune MD, Sparkes D, Curran MD, Zhang H, Jalal H. (2021)Specificity and positive predictive value of SARS-CoV-2 nucleic acid amplification testing in a low-prevalence setting. Clin Microbiol Infect. 27: 469.e9–469.e15. [View]
- Patra K. (2020) Around the NFL- All 77 false-positive COVID-19 tests come back negative upon reruns; August 24, 2020. [View]
- FDA. False positive results with BD SARS-CoV-2 reagents for the BD max system - letter to clinical laboratory staff and health care providers. Available from: https://www.fda.gov/medical-devices/letters-health-care-providers/false-positive-results-bd-sars-cov-2-reagents-bd-max-system-letter-clinical-laboratory-staff-and
- FDA. Risk of inaccurate results with thermo fisher scientific TaqPath COVID-19 combo kit - letter to clinical laboratory staff and health care providers. Available from: https://www.fda.gov/medical-devices/letters-health-care-providers/risk-inaccurate-results-thermo-fisher-scientific-taqpath-covid-19-combo-kit-letter-clinical?utm_campaign=2020-08-17%20Risk%20of%20Inaccurate%20Results%20with%20Thermo%20Fisher%20Scientific%20TaqPath&utm_medium=email&utm_source=Eloqua
- FDA. Potential for False Results with Roche Molecular Systems, Inc. cobas SARS-CoV-2 & Influenza Test for use on cobas Liat System-Letter to Clinical Laboratory Staff, Point-of-Care Facility Staff, and Health Care Providers. https://www.fda.gov/medical-devices/letters-health-care-providers/potential-false-results-roche-molecular-systems-inc-cobas-sars-cov-2-influenza-test-use-cobas-liat
- WHO Information Notice for IVD Users-Nucleic acid testing (NAT) technologies that use real-time polymerase chain reaction (RT-PCR) for detection of SARS-CoV-2 https://www.who.int/news/item/14-12-2020-who-information-notice-for-ivd-users
- WHO Genomic sequencing of SARS-CoV-2-A guide to implementation for maximum impact on public health. 8 January 2021. https://www.who.int/publications/i/item/9789240018440
- Bustin S, Mueller R, Shipley G, Nolan T. (2021) COVID-19 and Diagnostic Testing for SARS-CoV-2 by RT-qPCR-Facts and Fallacies. Int J Mol Sci. 22: 2459. [View]
- Anna MacDonald (2021) RT-qPCR—Facts and Fallacies: An Interview with Professor Stephen Bustin. Diagnostics from Technology Networks.[View]
- Borger P, Malhotra BR, Yeadon M, Craig C, McKernan K, Steger K.et al External peer review of the RTPCR test to detect SARS-CoV-2 reveals 10 major scientific flaws at the molecular and methodological level: consequences for false positive results. [View]
- Appenzeller T (1990) Democratizing the DNA sequence. Science 247: 1030–1032. [View]
- Rose TM, Schultz ER, Henikoff JG, Pietrokovski S, McCallum CM, Henikoff S (1998) Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. Nucleic Acids Res. 26: 1628–1635. [View]
- Garhyan J, Gharaibeh RZ, McGee S, Gibas CJ. (2013) The illusion of specific capture: surface and solution studies of suboptimal oligonucleotide hybridization. BMC Res Notes. 6: 72. [View]
- Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, et al. (2020) Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill 25: 2000045. [View]
- China National Center for Bioinformation. 2019 Novel Coronavirus Resource (2019nCoVR). https://ngdc.cncb.ac.cn/ncov//
- Facciponte DN, Bough MW, Seidler D, Carroll JL, Ashare A,et al. (2018) Identifying aerosolized cyanobacteria in the human respiratory tract: A proposed mechanism for cyanotoxin-associated diseases. Sci Total Environ. 645: 1003–1013. [View]
- Higuchi R, Fockler C, Dollinger G, Watson R. (1993) Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology (N Y). 11: 1026–1030. [View]
- Svec D, Tichopad A, Novosadova V, Pfaffl MW, Kubista M. (2015) How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomol Detect Quantif. 3: 9–16. [View]
- Mackay IM, Arden KE, Nitsche A. (2020) Real-time PCR in virology. Nucleic Acids Res. 30: 1292–1305. [View]
- Azadeh M, Gorovits B, Kamerud J, MacMannis S, Safavi A, Sailstad J, Sondag P. (2017) Calibration Curves in Quantitative Ligand Binding Assays: Recommendations and Best Practices for Preparation, Design, and Editing of Calibration Curves. AAPS J.20: 22. [View]
- Lee SH. (2020)Testing for SARS-CoV-2 in cellular components by routine nested RT-PCR followed by DNA sequencing. Int J Geriatr Rehabil. 2: 69–96. [View]
- https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/faqs-testing-sars-cov-2
- https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas
- Ryu KH, Choi SH, Lee JS. (2000) Restriction primers as short as 6-mers for PCR amplification of bacterial and plant genomic DNA and plant viral RNA. Mol Biotechnol. 14: 1–3. [View]
- Lee SH. Toll-like receptor 9 agonists in HPV vaccine Gardasil9. International Journal of Vaccine Theory, Practice, and Research. 2021; 1: 295-317. https://ijvtpr.com/index.php/IJVTPR/article/view/13/21
- Kwok S, Kellogg DE, McKinney N, Spasic D, Goda L, Levenson C, Sninsky JJ. (1990)Effects of primer-template mismatches on the polymerase chain reaction: : human immunodeficiency virus type 1 model studies. Nucleic Acids Res. 18: 999–1005. [View]
- O’Dell SD, Humphries SE, Day IN. (1996) PCR induction of a TaqI restriction site at any CpG dinucleotide using two mismatched primers (CpG-PCR). Genome Res. 6: 558–68. [View]
- Ruiz-Villalba A, van Pelt-Verkuil E, Gunst QD, Ruijter JM, van den Hoff MJ. (2017) Amplification of nonspecific products in quantitative polymerase chain reactions (qPCR). Biomol Detect Quantif. 14: 7–18. [View]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M,et al. (2009)The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 55: 611–22. [View]
- FDA. Nucleic Acid Amplification Assay for the Detection of Enterovirus RNA - Class II Special Controls Guidance for Industry and FDA Staff. https://www.fda.gov/medical-devices/guidance-documents-medical-devices-and-radiation-emitting-products/nucleic-acid-amplification-assay-detection-enterovirus-rna-class-ii-special-controls-guidance
- FDA. Establishing the Performance Characteristics of In Vitro Diagnostic Devices for the Detection or Detection and Differentiation of Human Papillomaviruses. https://www.fda.gov/media/92930/download
- Challen R, Brooks-Pollock E, Read JM, Dyson L, Tsaneva-Atanasova K, Danon L. (2002) Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: matched cohort study. BMJ. 372: n579. [View]
- Applied Biosystems. Protocol for sequencing the SARS-CoV-2 S gene 69-70del. https://assets.thermofisher.com/TFS-Assets/GSD/brochures/sequencing-sars-cov-2-s-gene-69-70del-protocol.pdf
- Nowakowski J, Schwartz I, Liveris D, Wang G, Aguero-Rosenfeld ME, Girao G,et al.(2001) Lyme Disease Study Group. Laboratory diagnostic techniques for patients with early Lyme disease associated with erythema migrans: a comparison of different techniques. Clin Infect Dis 33: 2023–2027. [View]
- Nelson, Christina A., MD, MPH. CDC Expert Commentary, June 2012. PCR for Diagnosis of Lyme Disease: Is It Useful? https://www.cdc.gov/lyme/healthcare/index.html
- Schiffman M, de Sanjose S (2019) False positive cervical HPV screening test results. Papillomavirus Res 7: 184–187. [View]
- Schneider, E. Analysis Summary Report. New York State Department of Health Evaluation of the Human Papilloma Virus (HPV) Proficiency Test from October 2015.
- Stout NK, Goldhaber-Fiebert JD, Ortendahl JD, Goldie SJ (2008) Trade-offs in cervical cancer prevention: balancing benefits and risks. Arch Intern Med 168: 1881–1889. [View]
- CAP Today. Anne Paxton. Primary HPV screen only? Experts warn of risks. January issue, 2021. [View]
- The Irish Times. Emma Mhic Mhathúna settles cervical smear case for €7.5 million. [View]
- CDC (2021) COVID-19 Advisory. Nucleic Acid Amplification Tests (NAATs). Updated Mar. 24, 2021. [View]
Supplemental File
Nested PCR amplification followed by Sanger sequencing for detection of SARS-CoV-2 N gene in 30 nasopharyngeal swab specimens previously tested positive by two EUA RT-qPCR assays
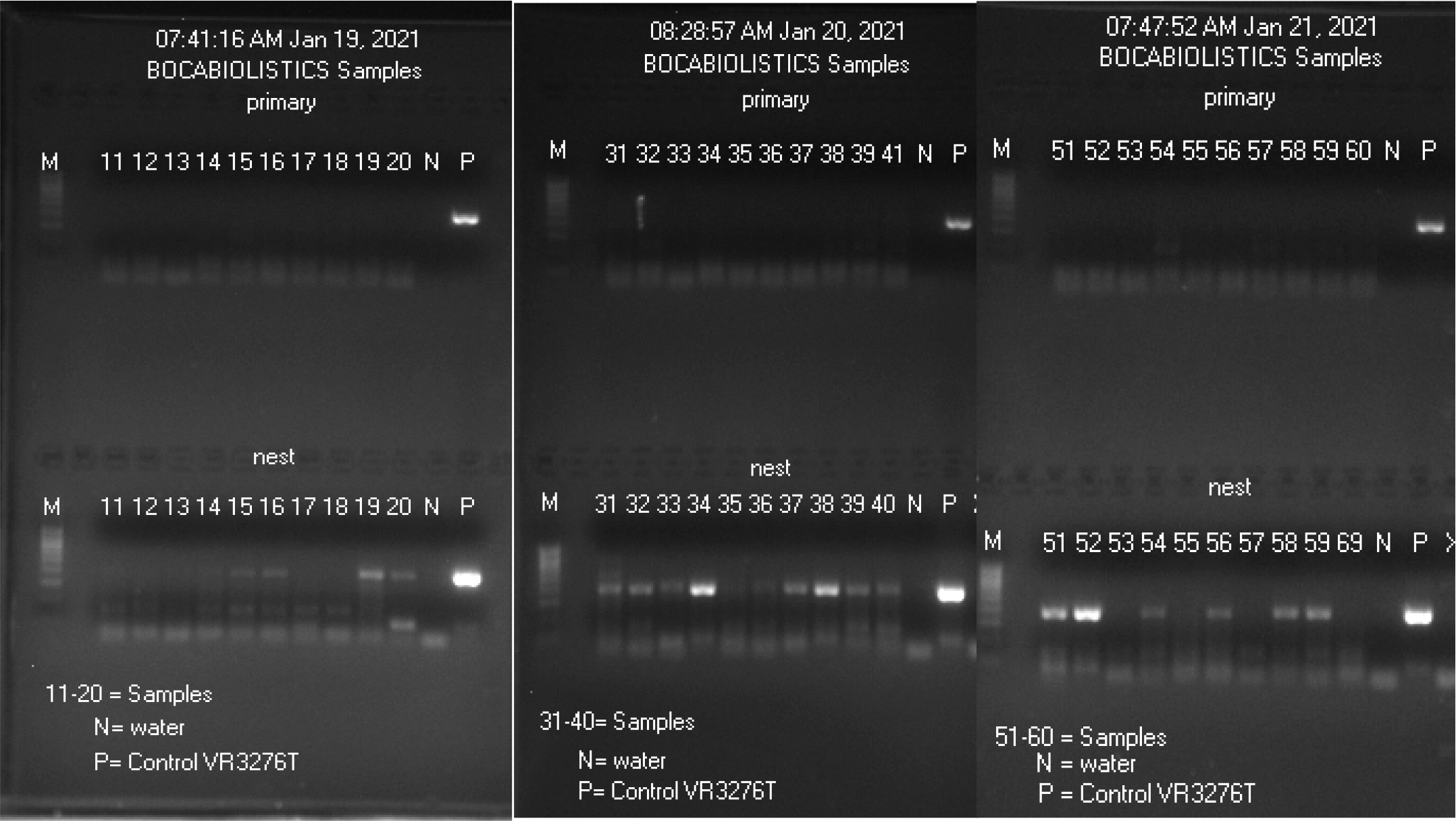
M21- 11-20, M21- 31-40 and M21- 51-60 were presumptive Positive samples.
- Images of gel electrophoresis of primary and nested PCR amplification of 30 RT-qPCR positive nasopharyngeal swab samples
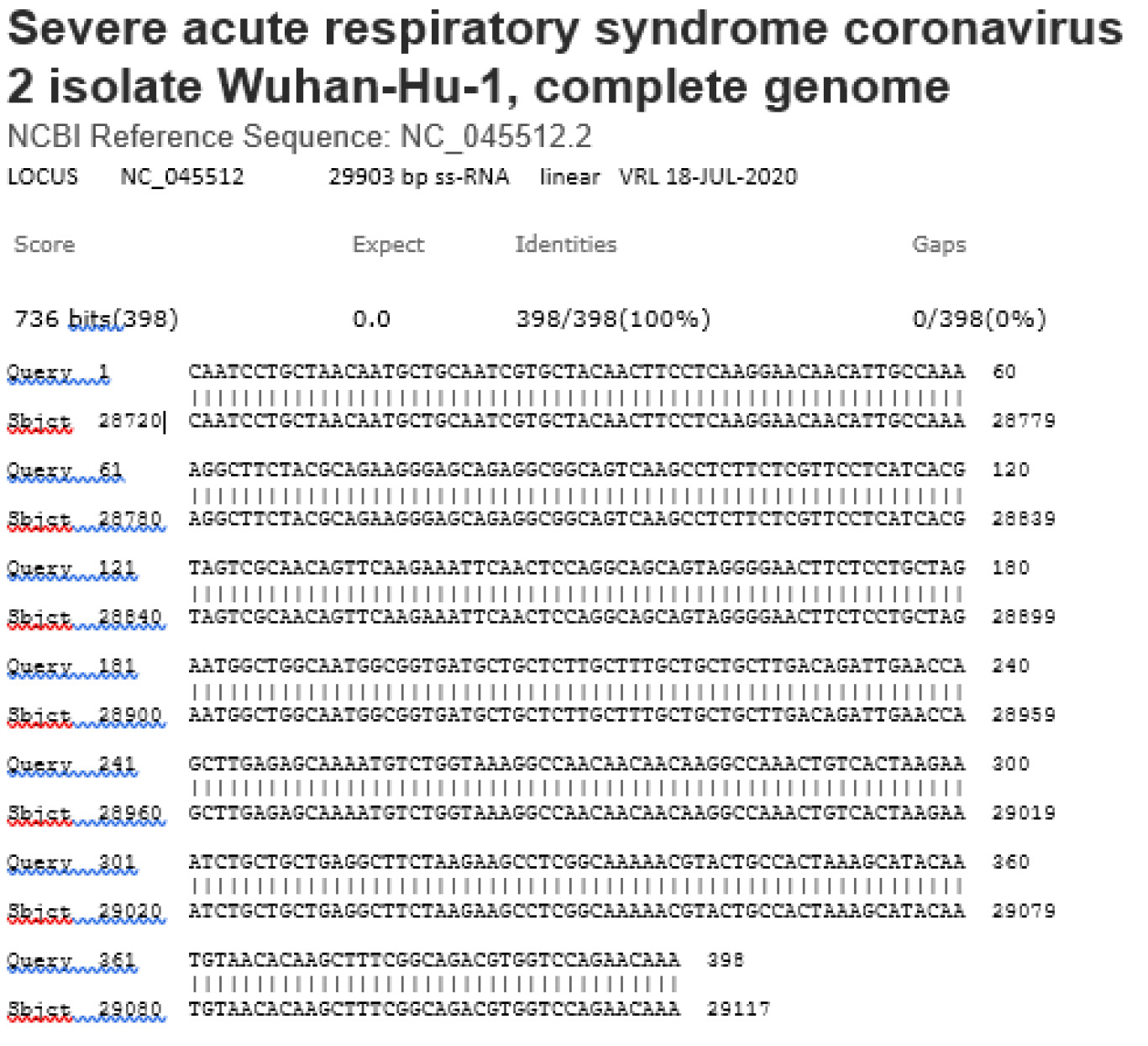
- Sanger sequencing of all nested PCR products derived from the 30 “positive” clinical samples (shown in lanes 11-20, 31-40 and 51-60) with Co4 forward primer showed a true positive SARS-CoV-2 N gene sequence in M21- samples #15, 16, 19, 20, 32, 34, 37, 38, 39, 40, 51, 52, 54, 56, 58, and 59. These 16 true-positive nested PCR products were re-sequenced with Co3 reverse primer for a bi-directional sequencing for single nucleotide mutation typing. After the bi-directional sequences were converted into 5’-3’ one-directional reading, the composite 398-base sequence was submitted to the GenBank for BLAST analysis. The report from the GenBank was copied and pasted below each positive pair of electropherograms. The sequencing data of the 30 “positive”, including the false-positive, samples are presented as follows. The PCR false-positive samples were labeled as “Negative sequencing”.
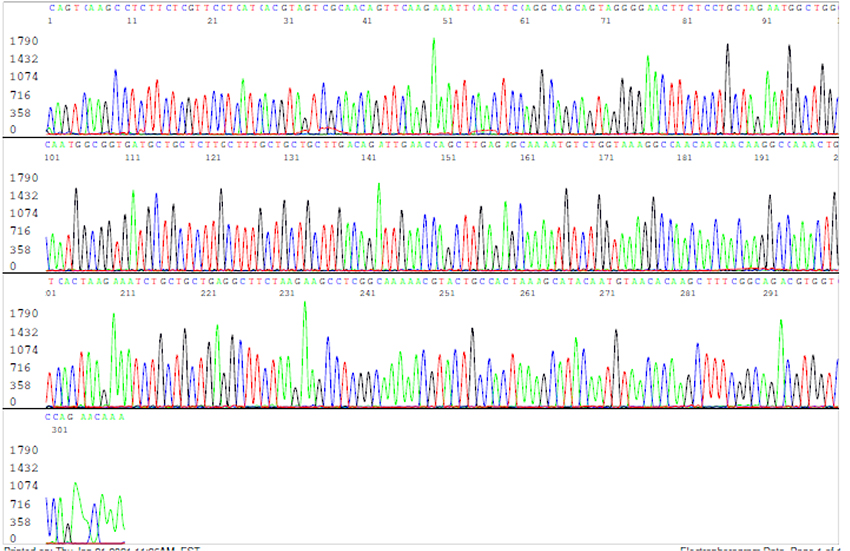
Two positive control bi-directional sequencing electropherograms of a 398-base N gene segment with its GenBank BLAST report of a SARS-CoV-2 Wuhan-Hu-1 prototype sequence are presented first.
Control Sanger sequencing with Co4 forward sequencing primer Wuhan-Hu-1 prototype
Control Sanger sequencing with Co3 reverse sequencing primer Wuhan-Hu-1 prototype
M21-11 Negative sequencing
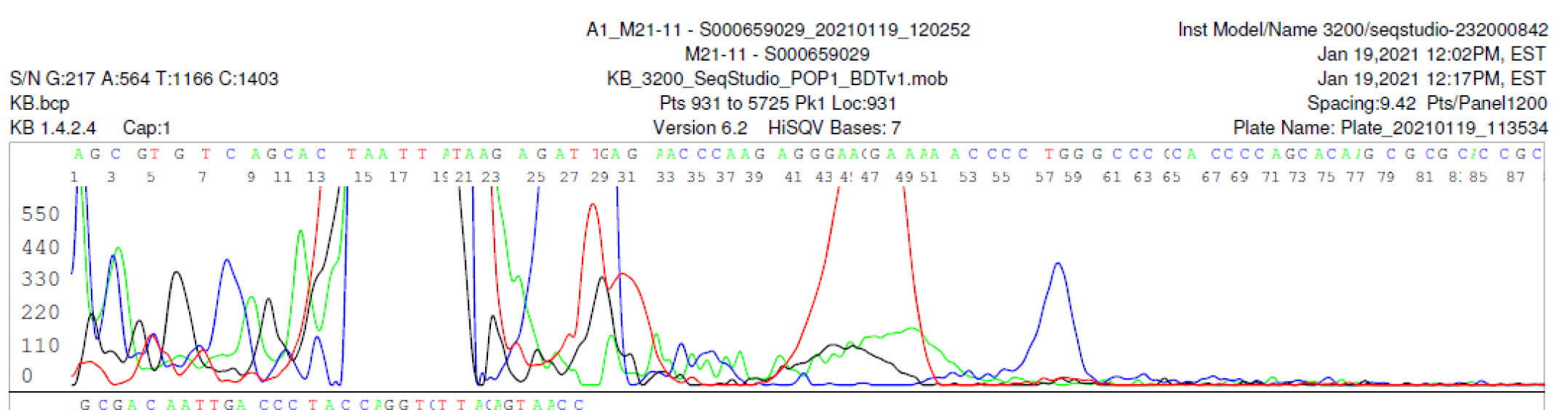
M21-12 Negative sequencing
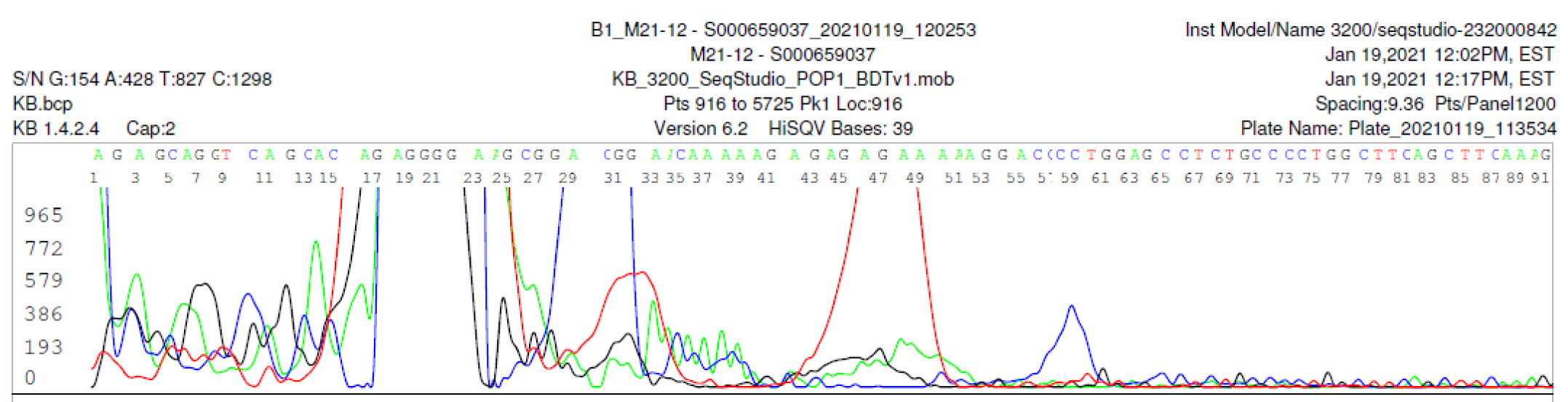
M21-13 Negative sequencing
M21-14 Negative sequencing
M21-15 Positive bi-directional sequencing and BLAST with 4 single nucleotide mutations typed in red
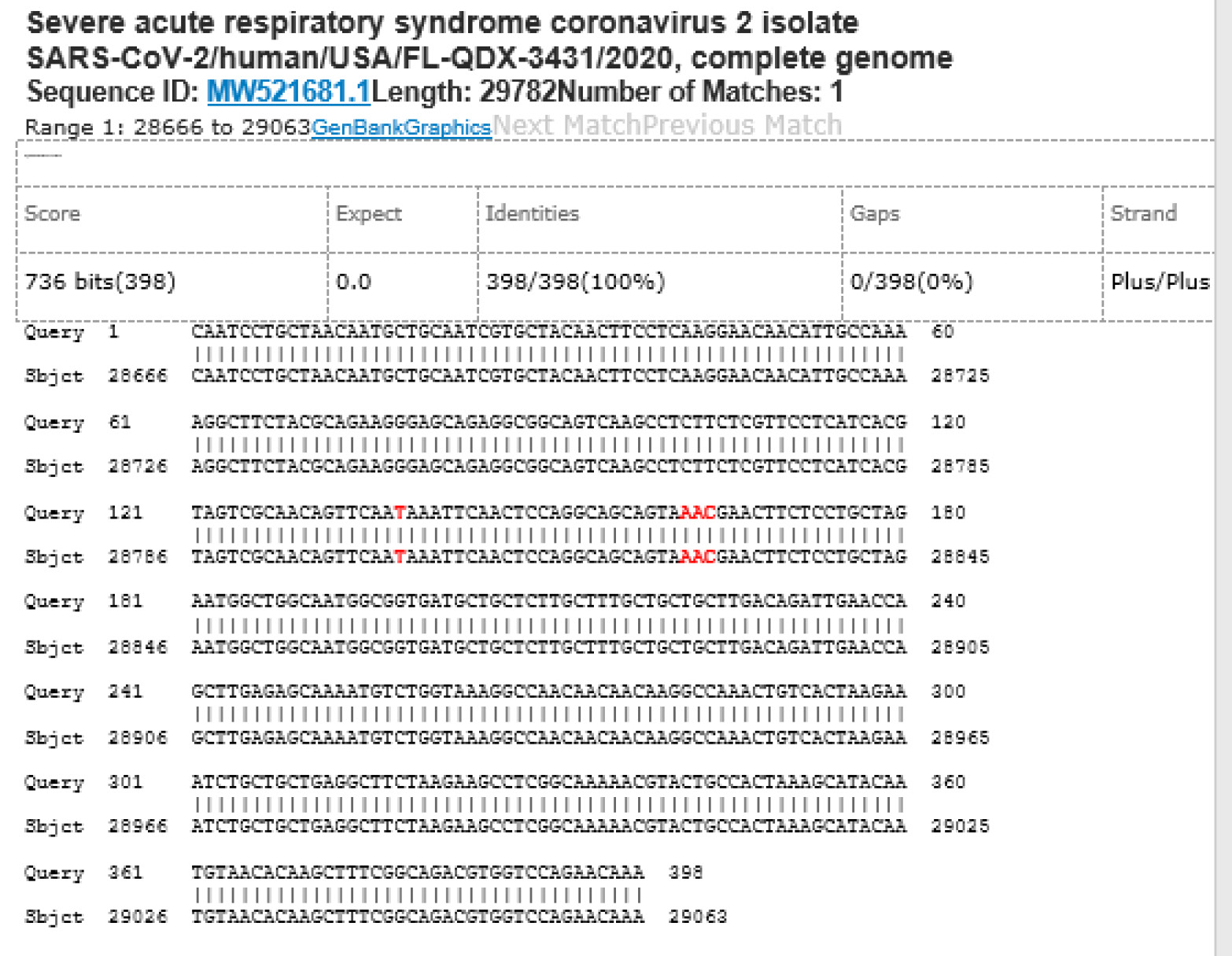
M21-16 Positive bi-directional sequencing and BLAST with 1 single nucleotide mutation typed in red
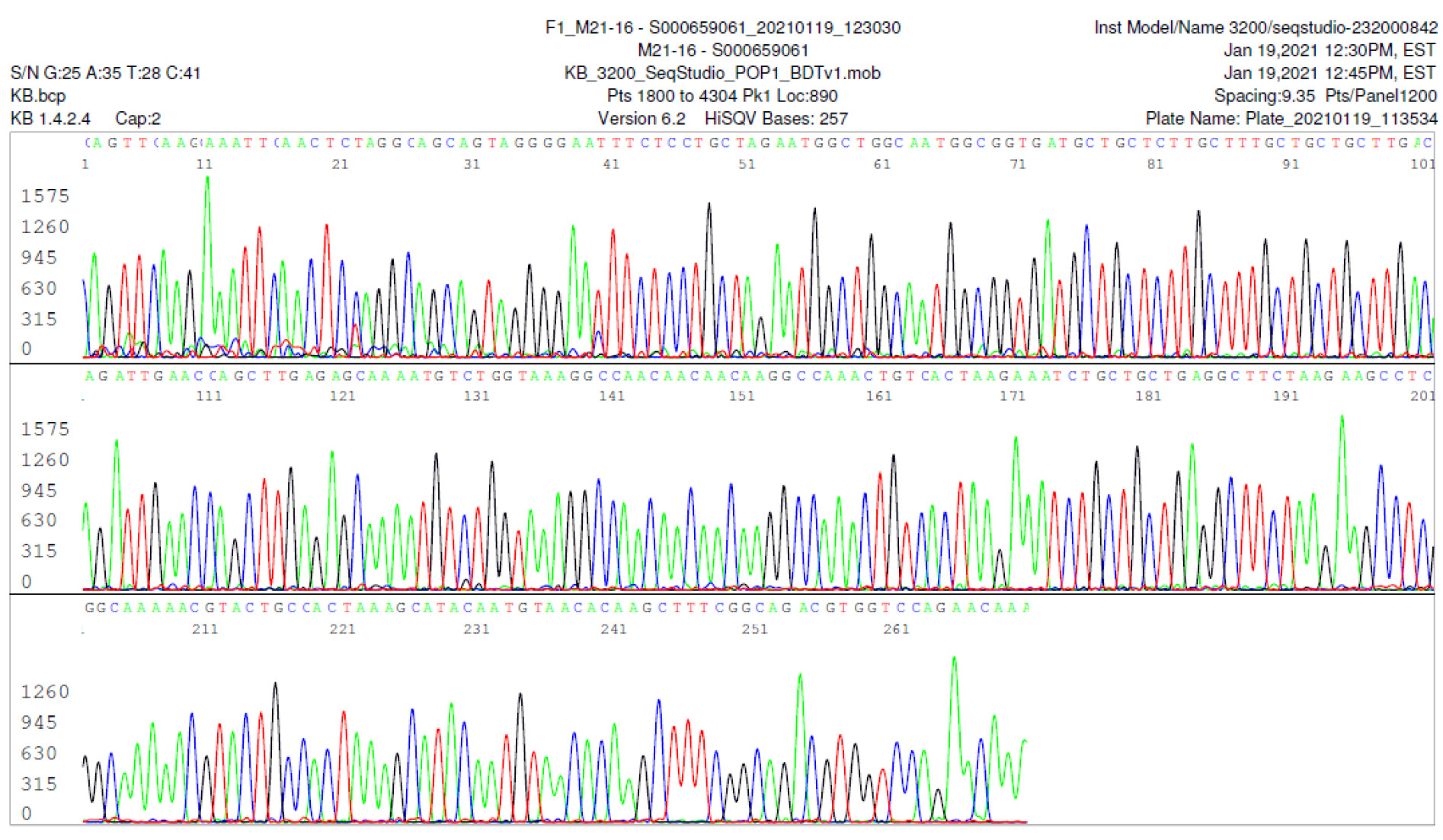
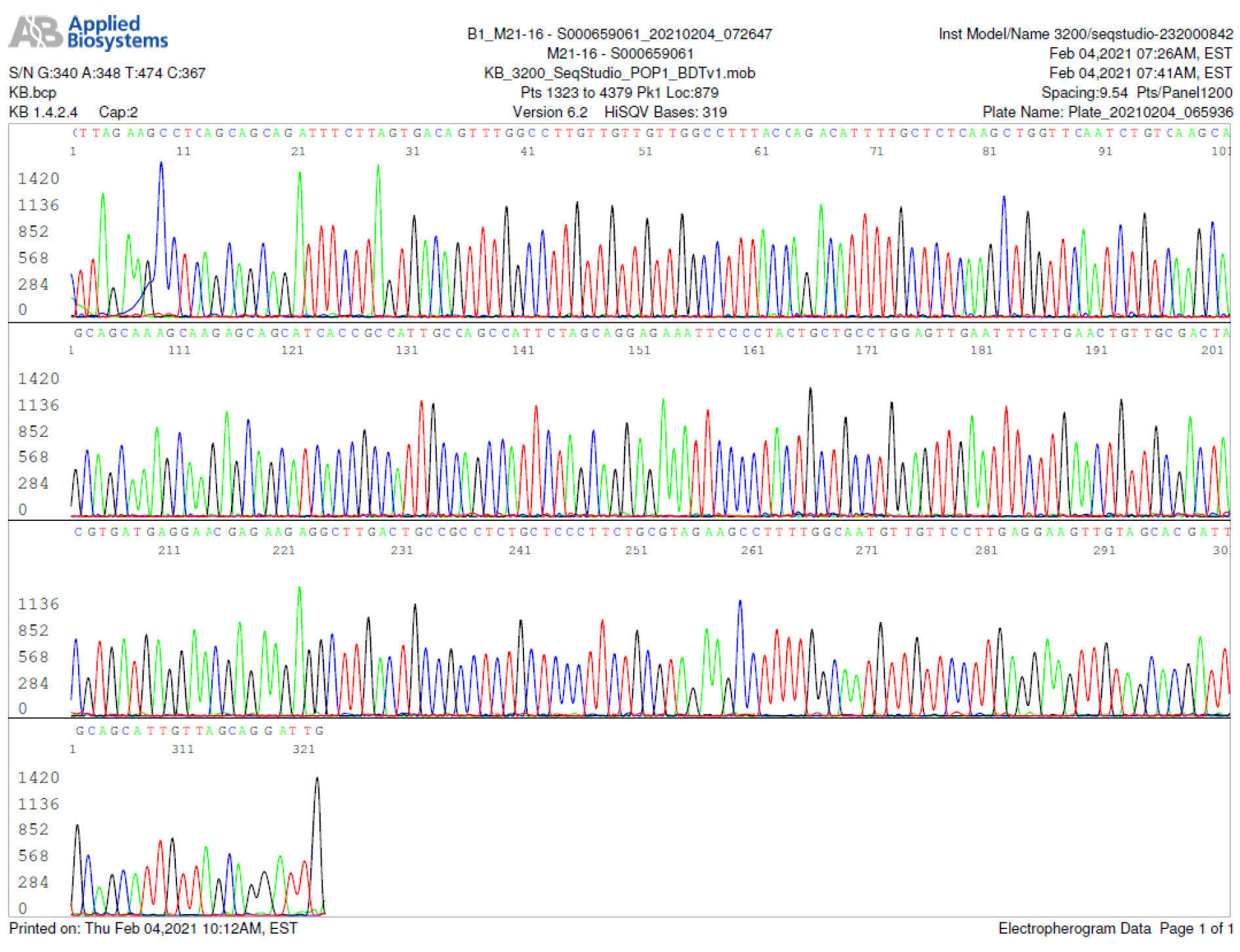
M21-17 Negative sequencing
M21-18 Negative sequencing
M21-19 Positive bi-directional sequencing and BLAST with 1 single nucleotide mutation typed in red
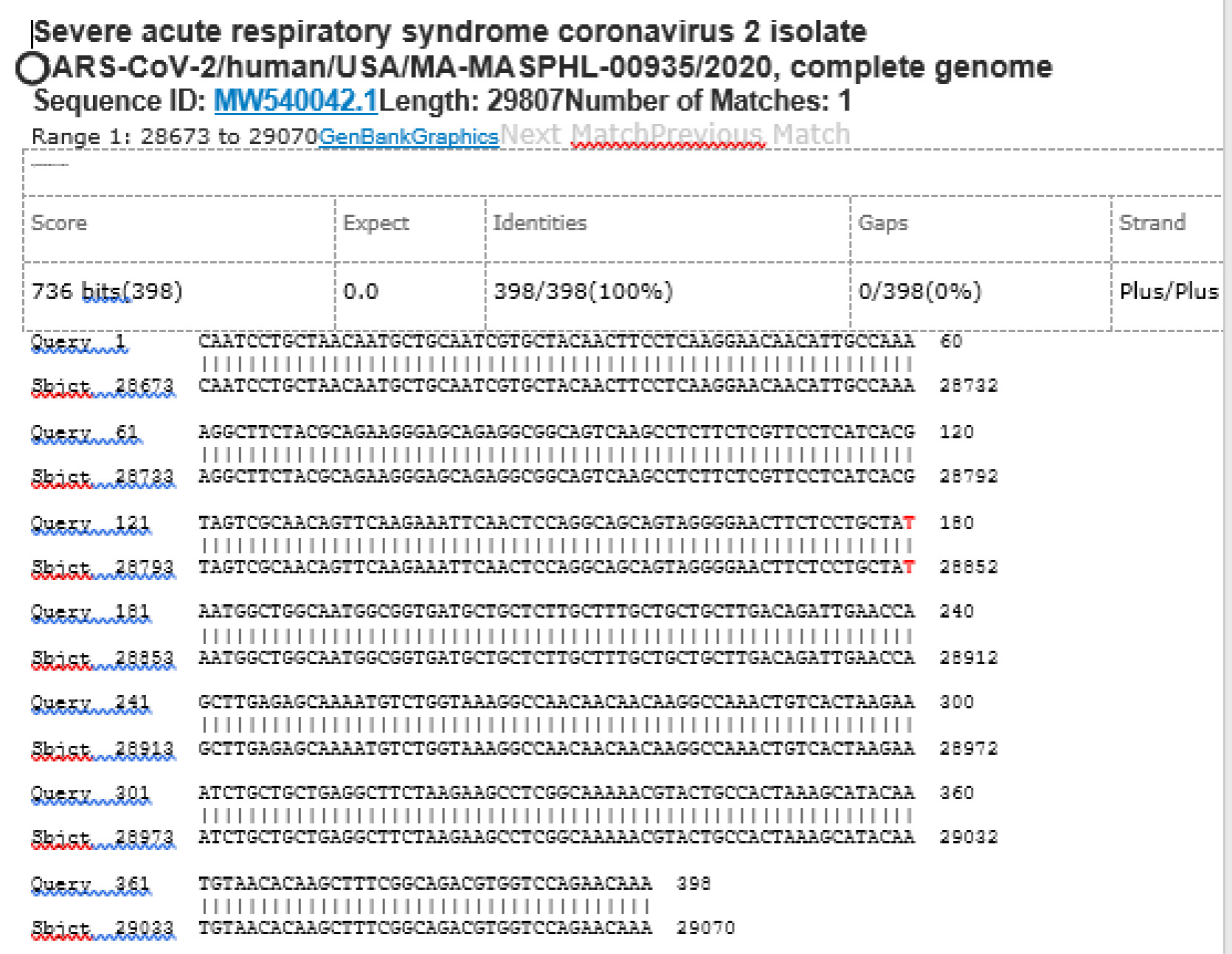
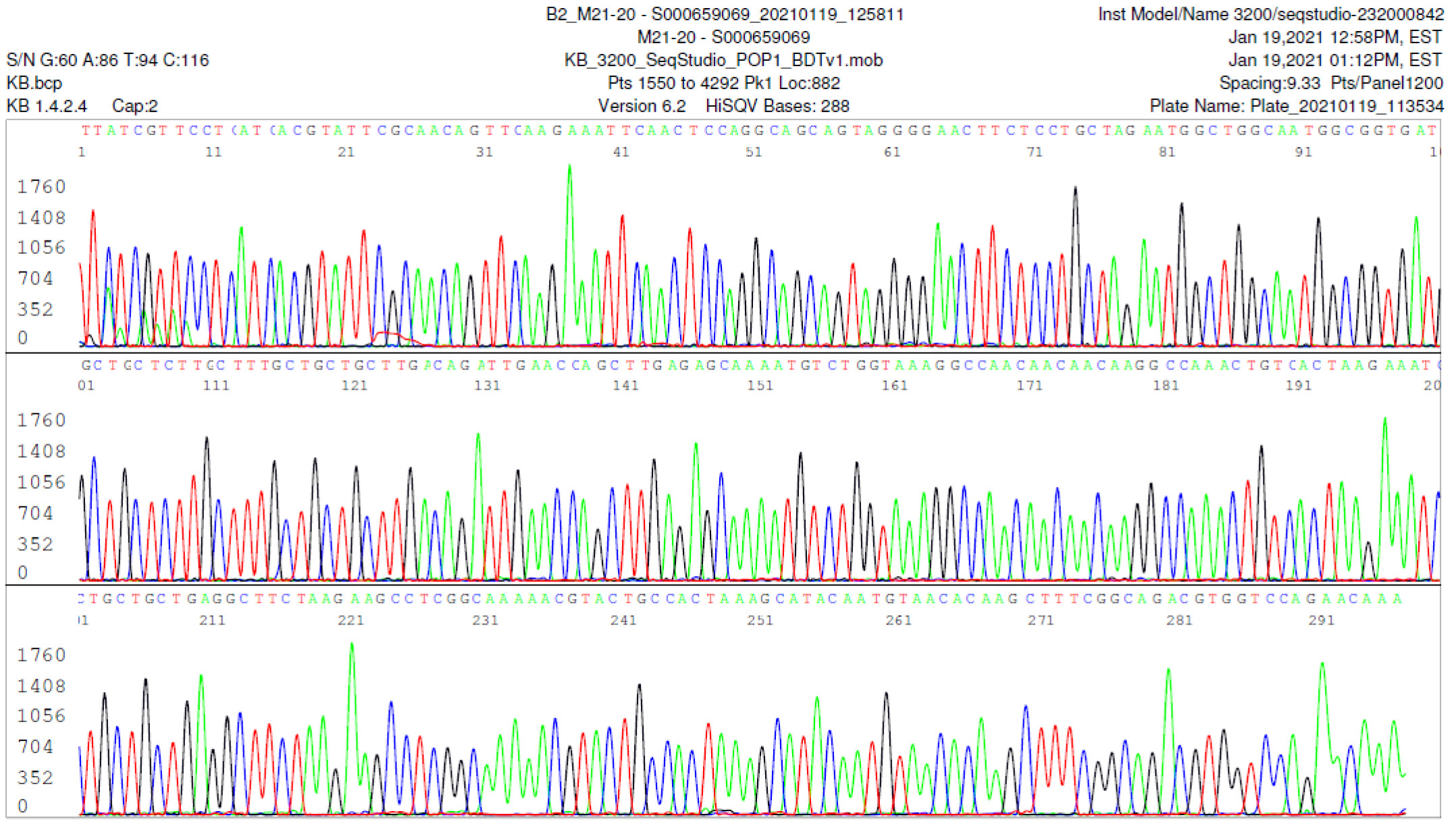
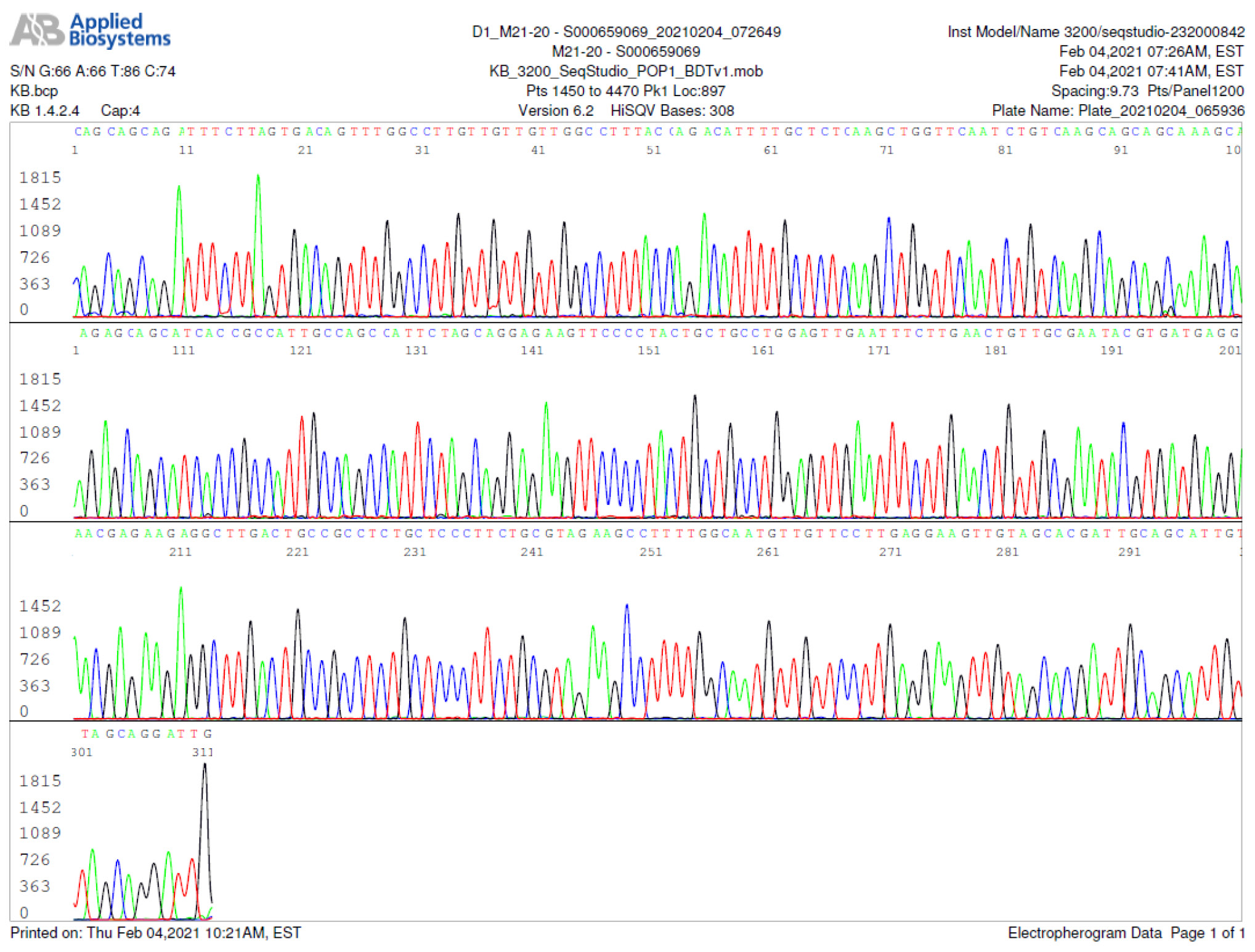
M21-20 Positive bi-directional sequencing and BLAST with 1 single nucleotide mutation typed in red
M21-31 Negative sequencing
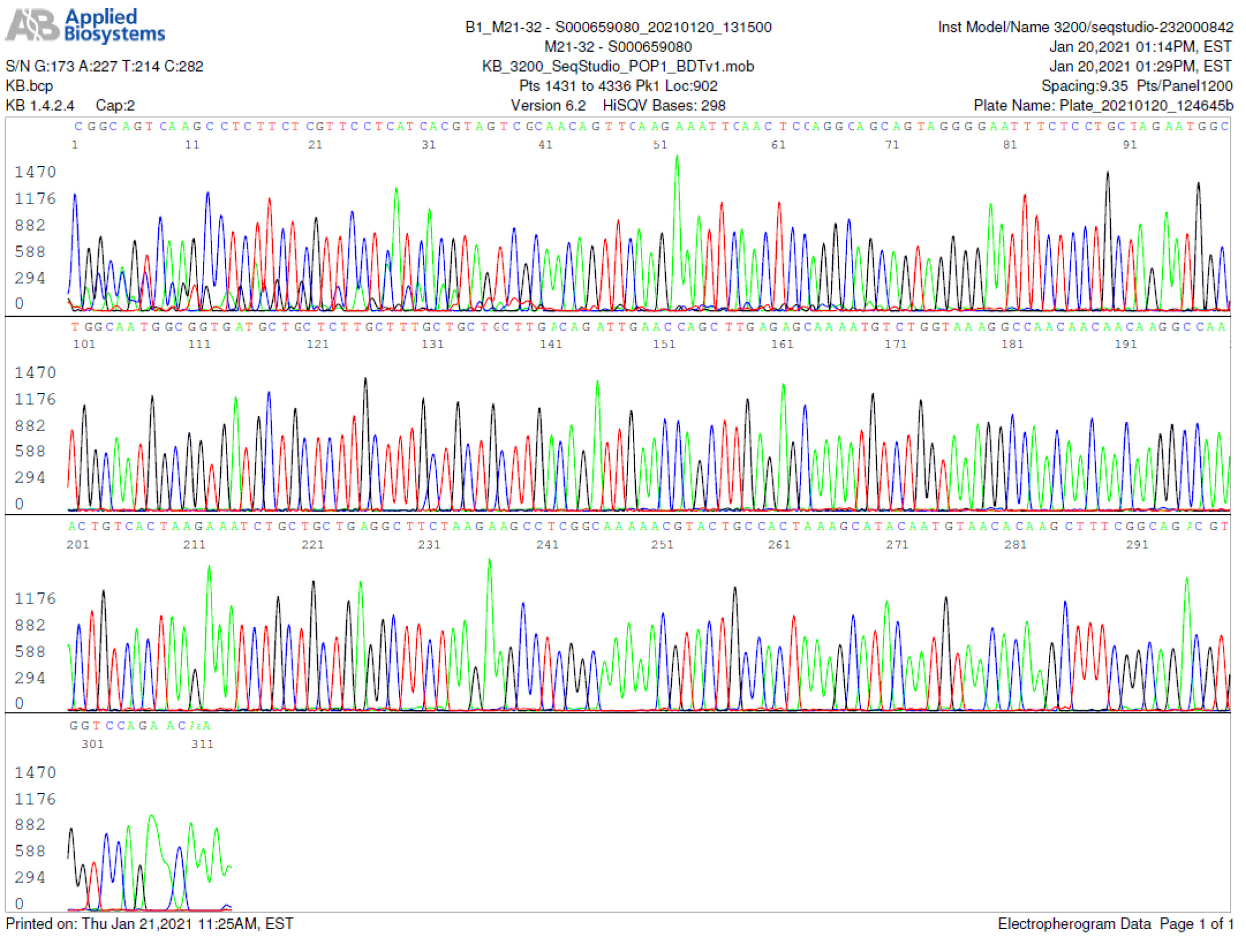
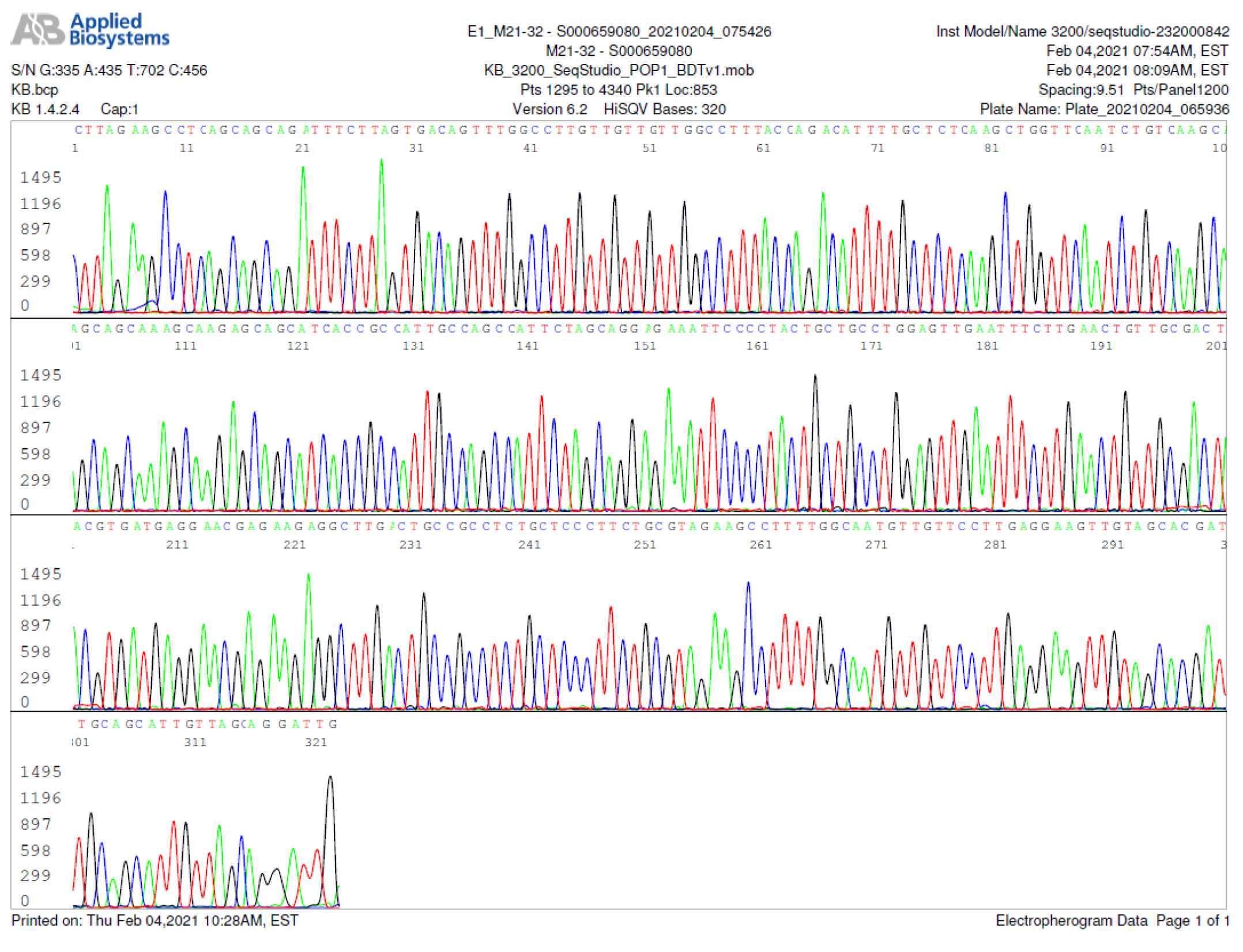
M21-32 Positive bi-directional sequencing and BLAST with 1 single nucleotide mutation typed in red

M21-33 Negative sequencing
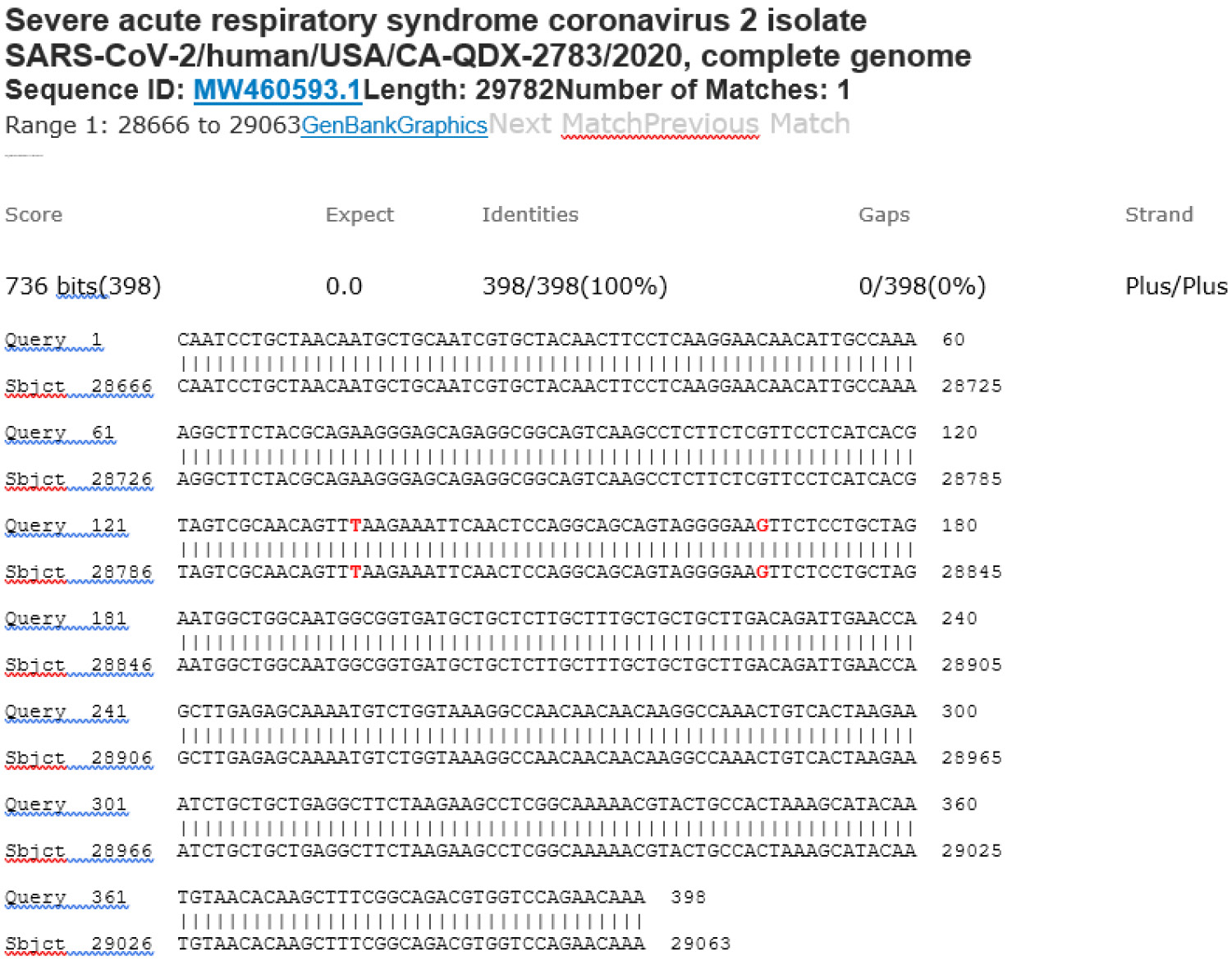
M21-34 Positive bi-directional sequencing and BLAST with 2 single nucleotide mutations typed in red
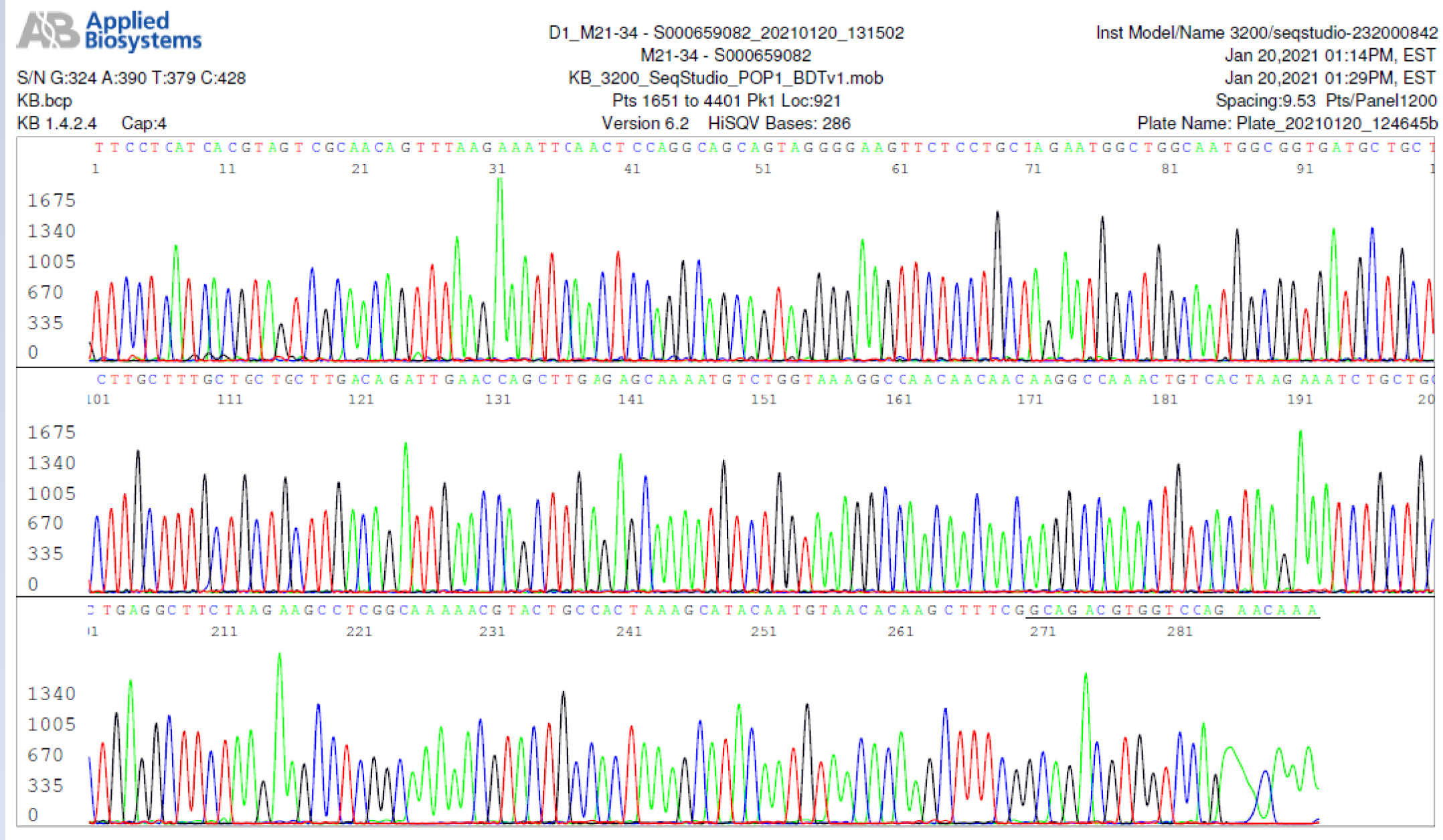
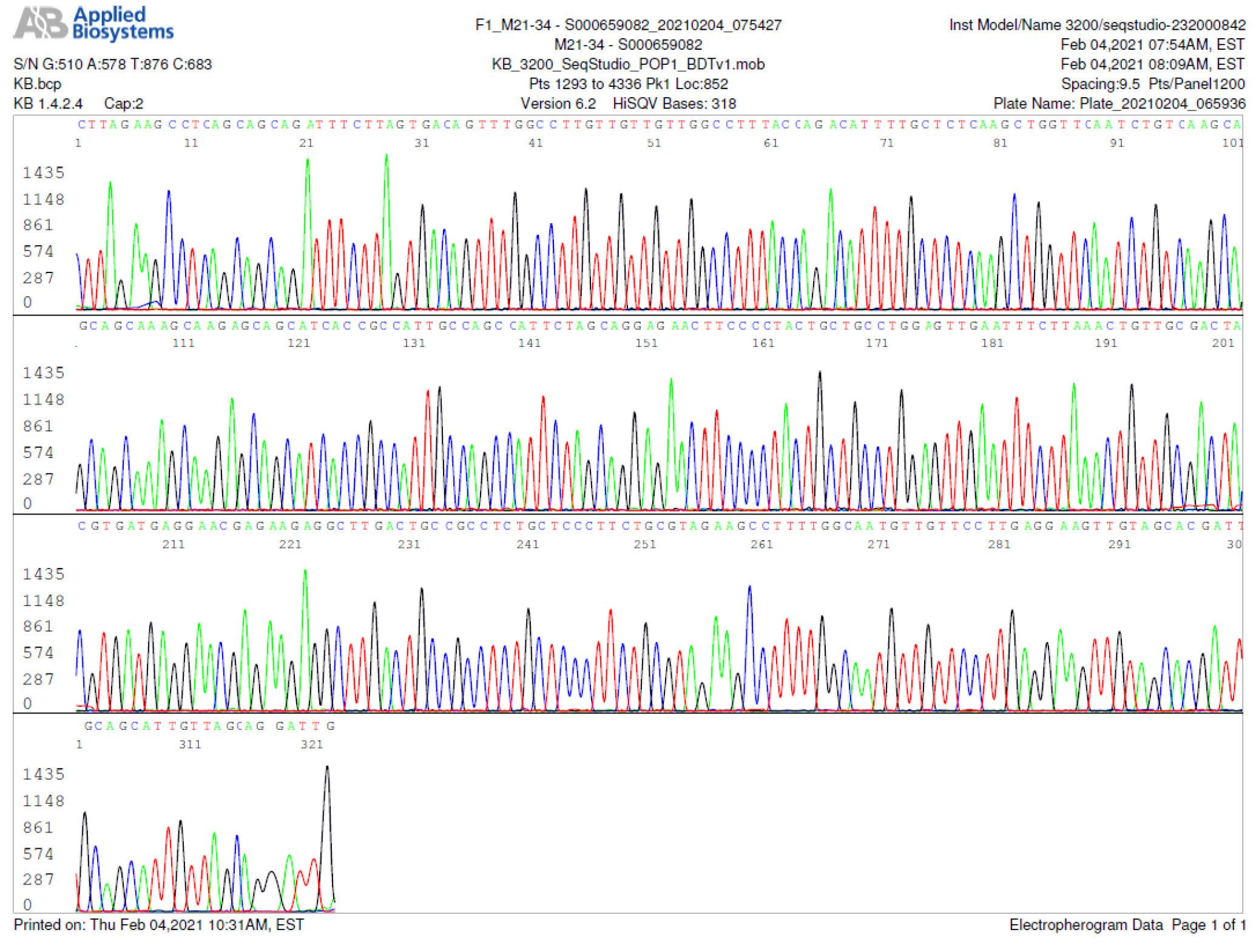
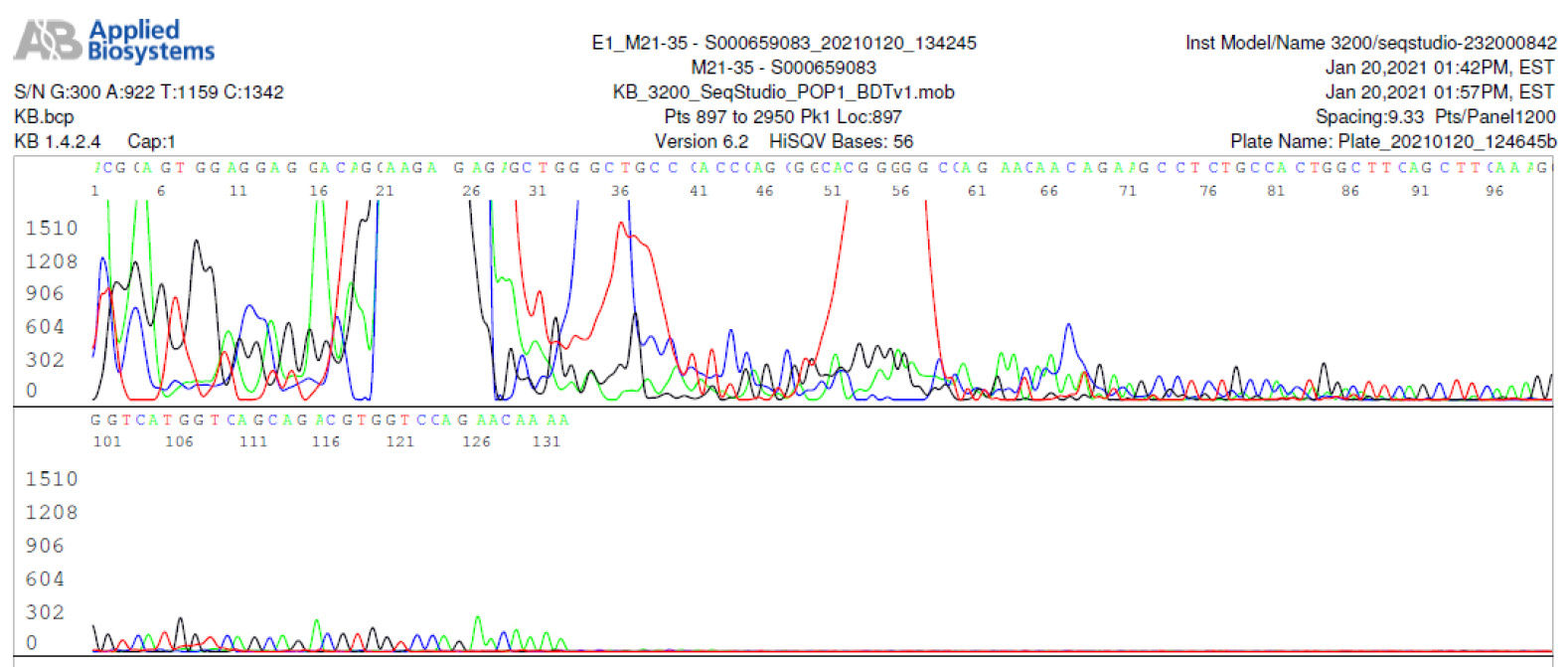
M21-35 Negative sequencing with human gene amplification
M21-36 Negative sequencing
M21-37 Positive bi-directional sequencing and BLAST with 1 single nucleotide mutation typed in red

M21-38 Positive bi-directional sequencing and BLAST with 2 single nucleotide mutations typed in red
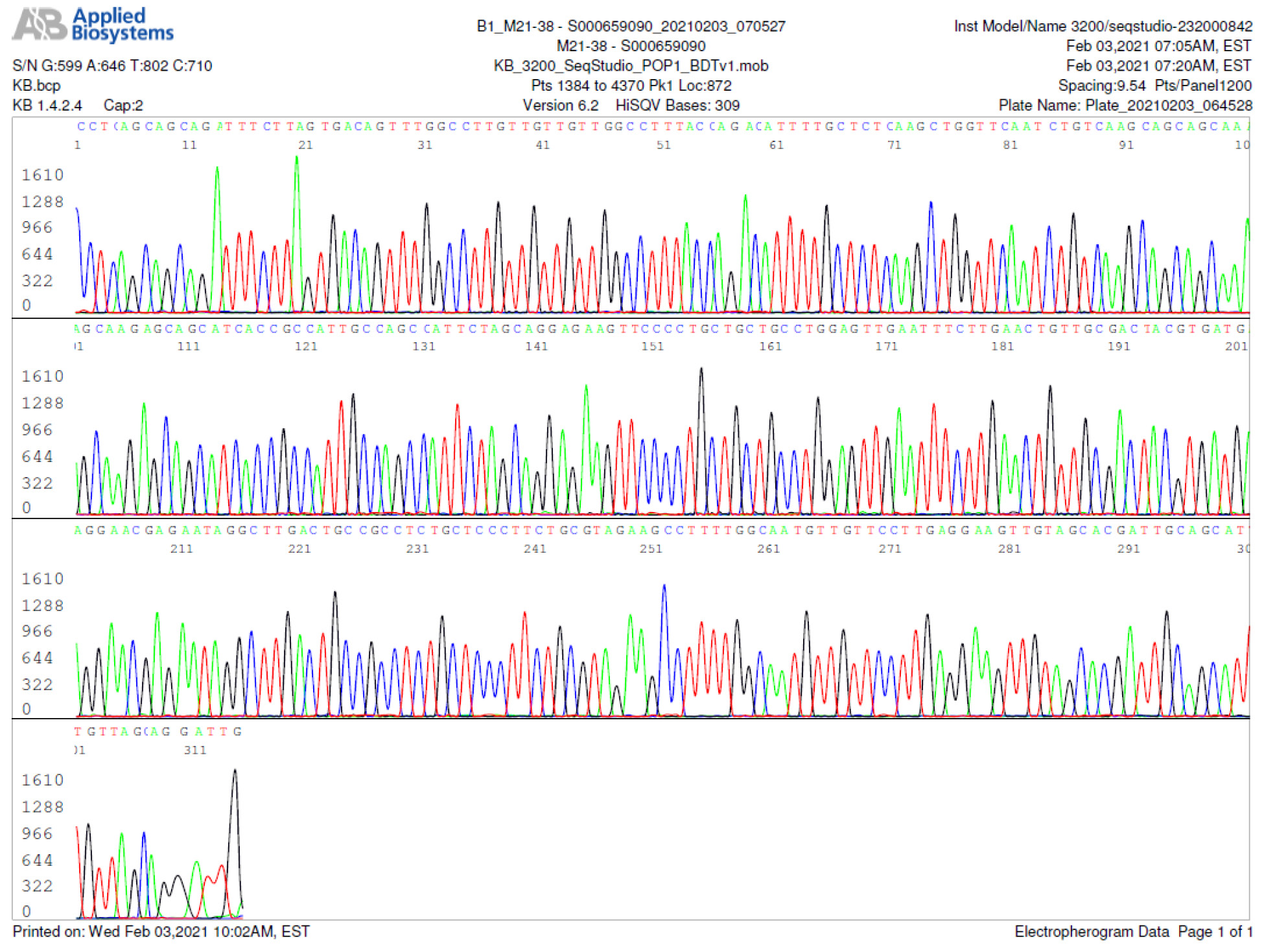
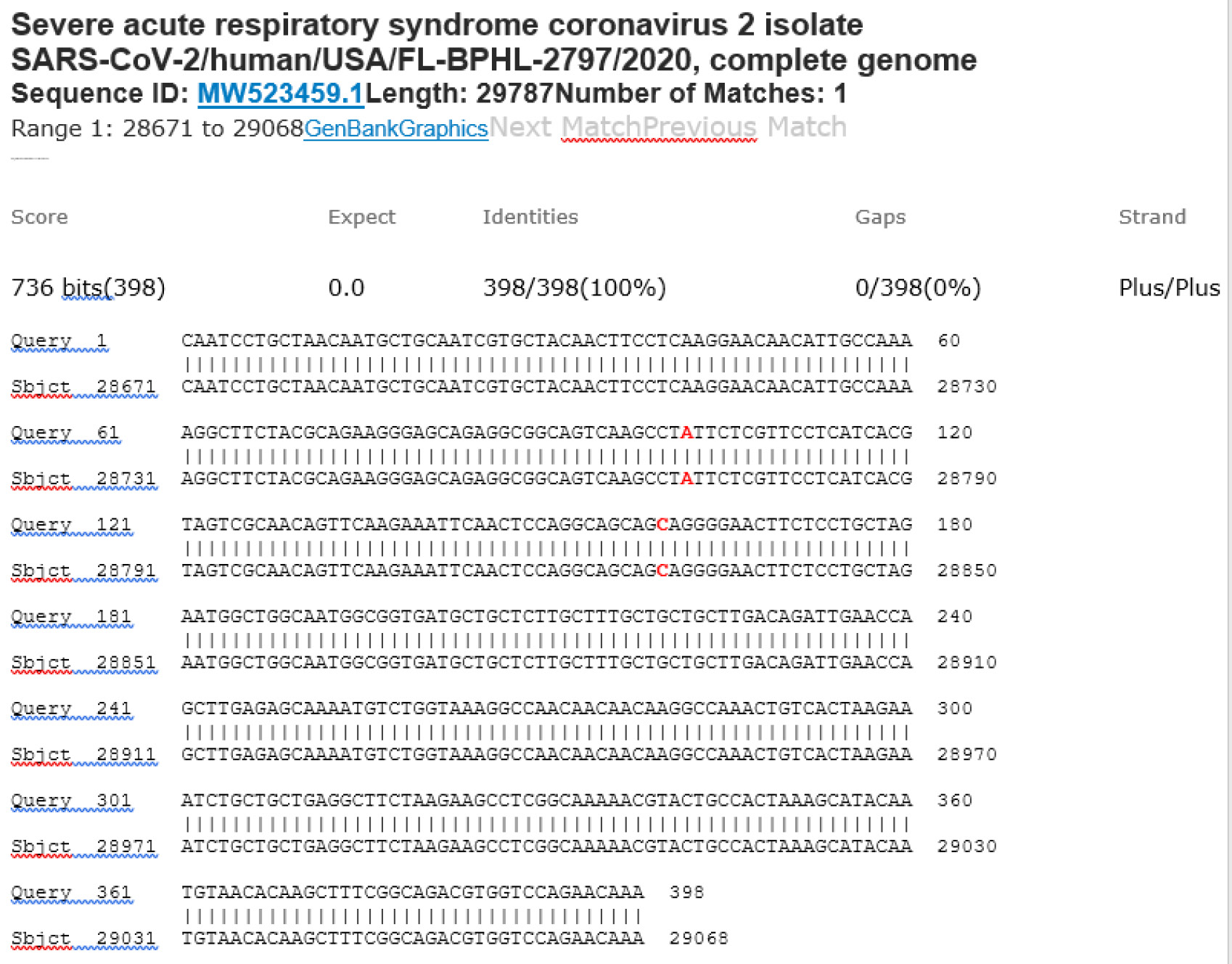
M21-39 Positive bi-directional sequencing and BLAST with 3 single nucleotide mutations typed in red
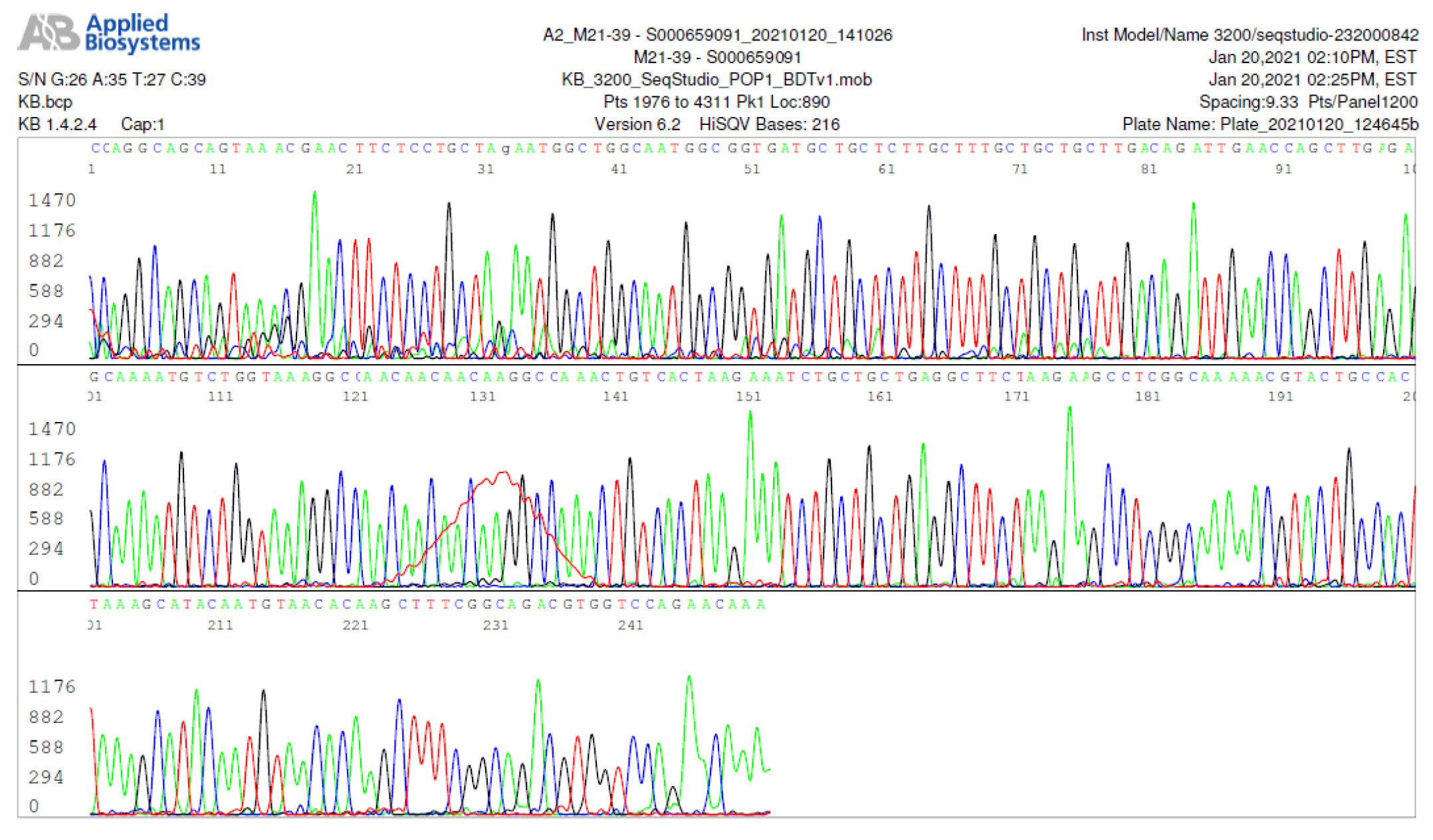
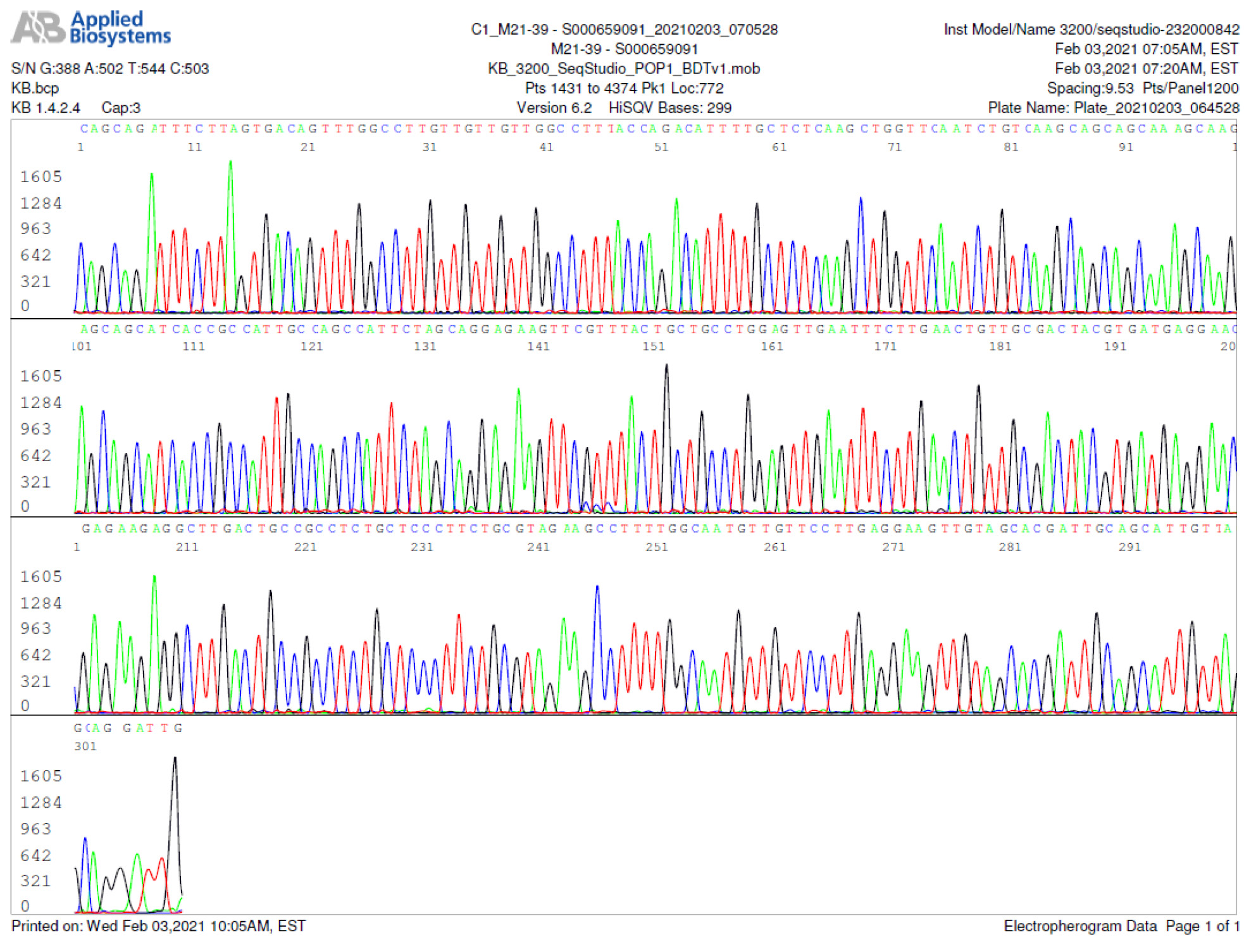
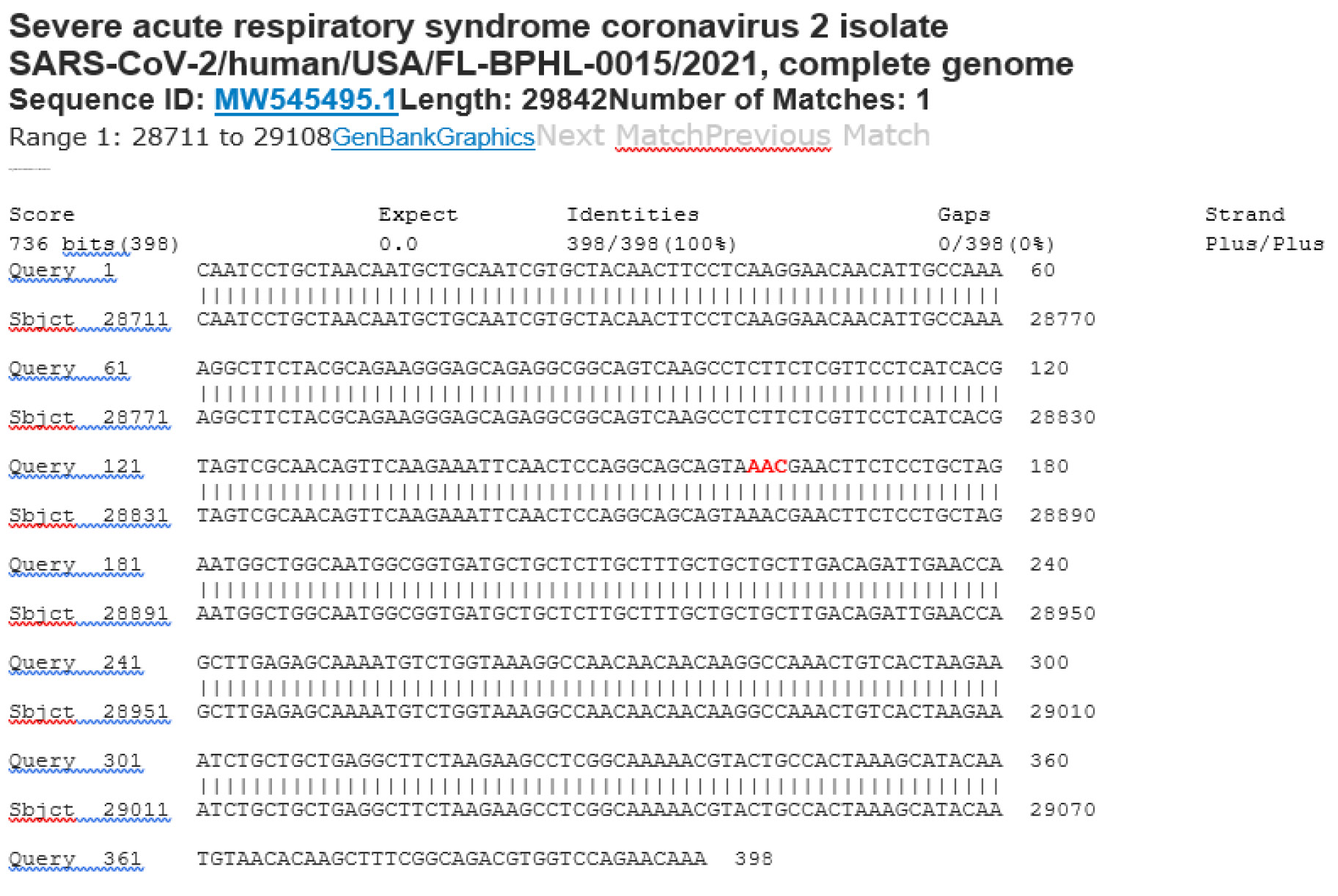
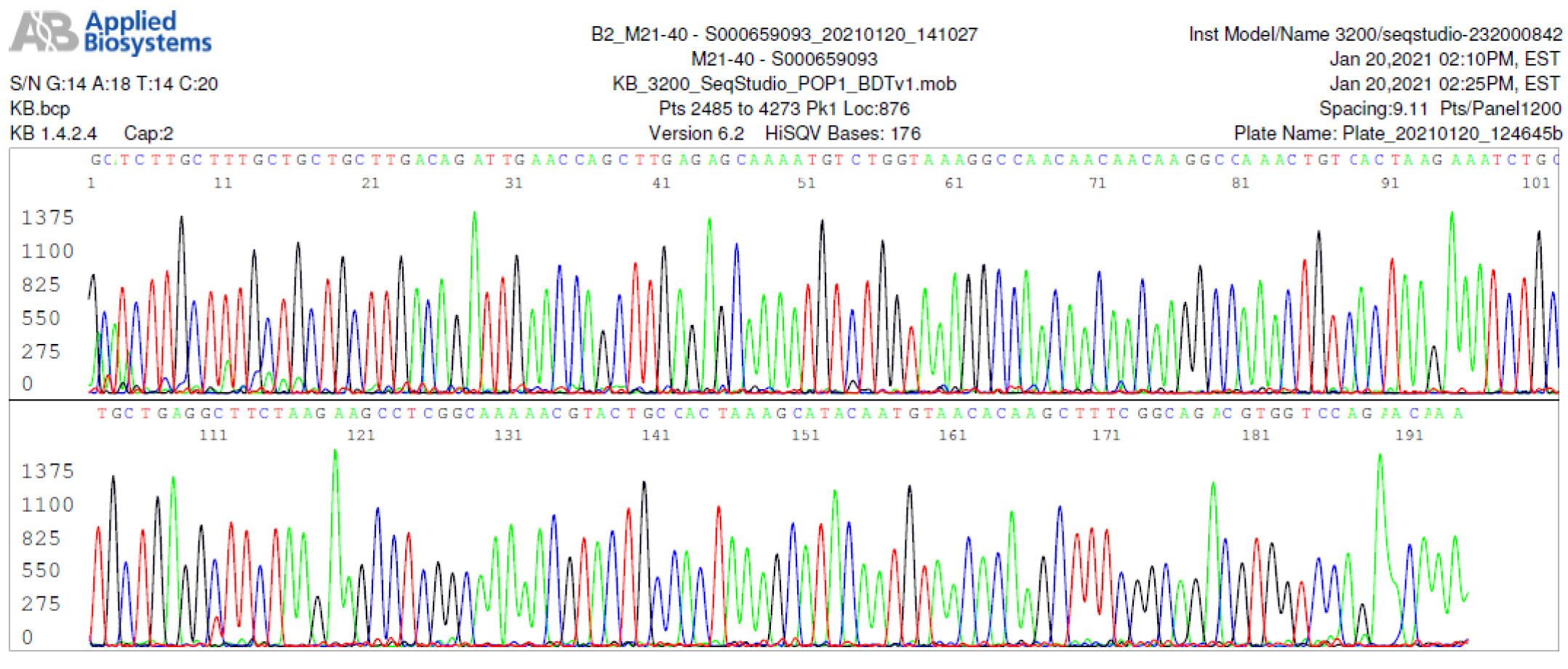
M21-40 Positive bi-directional sequencing and BLAST with 1 single nucleotide mutation typed in red
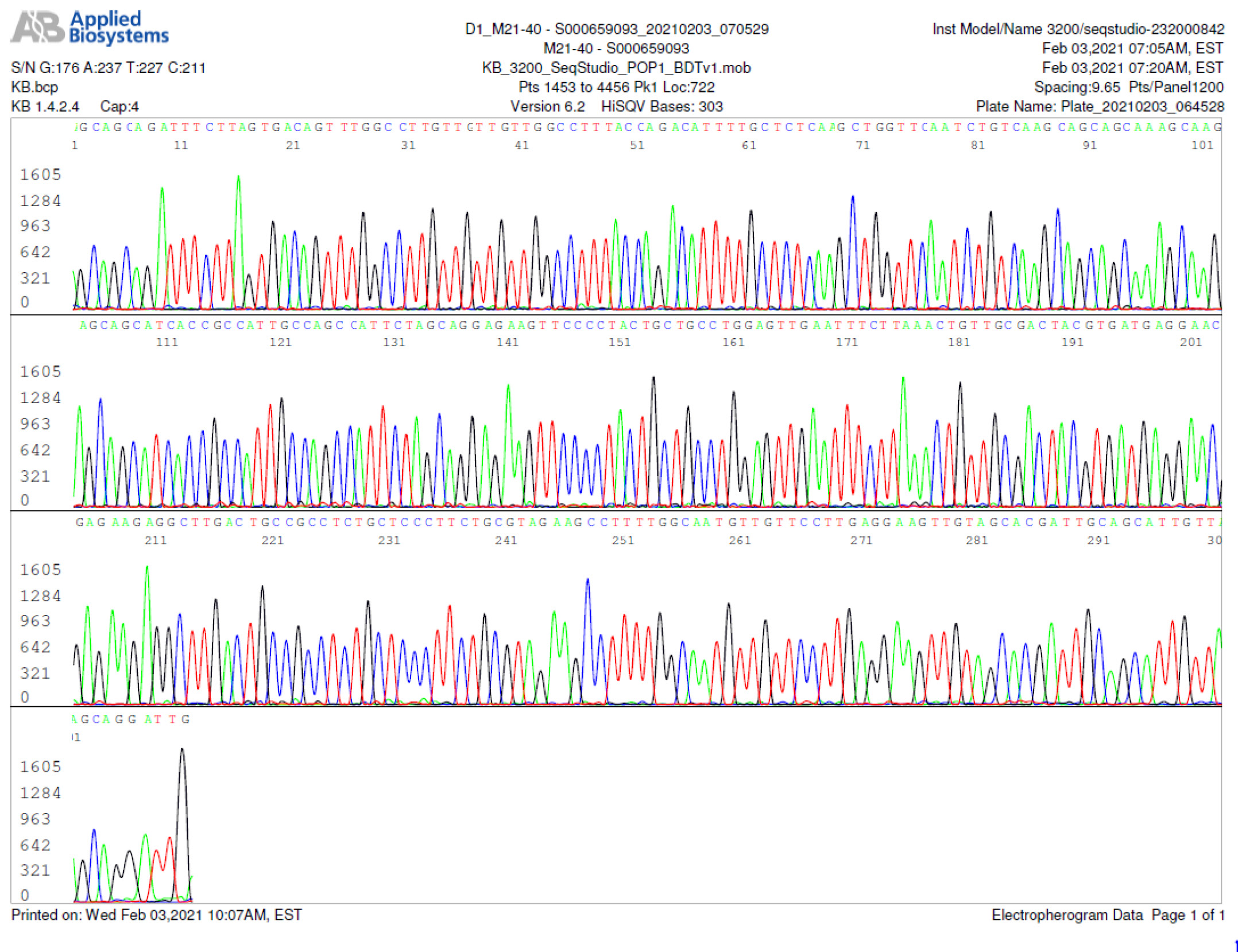

M21-51 Positive bi-directional sequencing and BLAST with 1 single nucleotide mutation typed in red
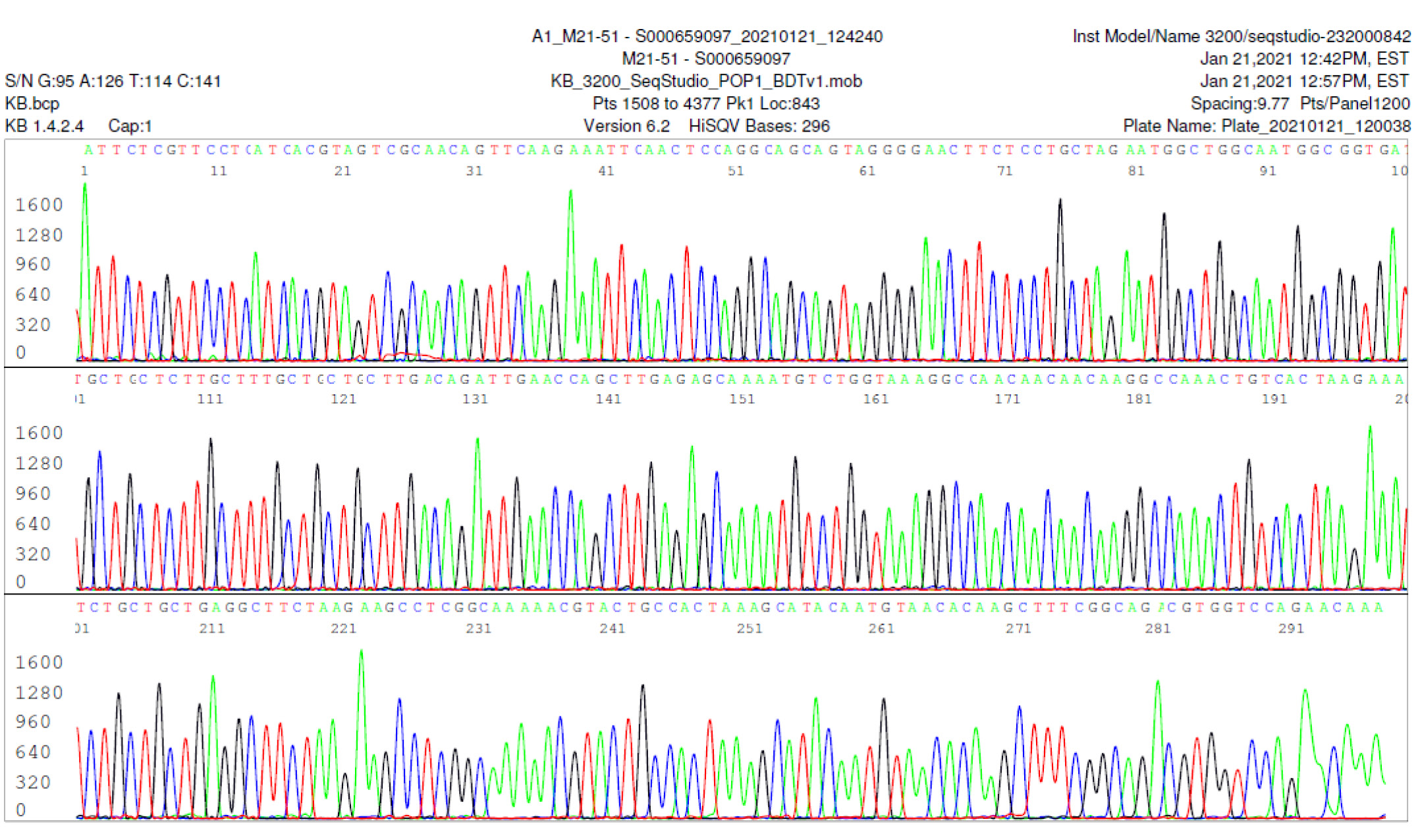
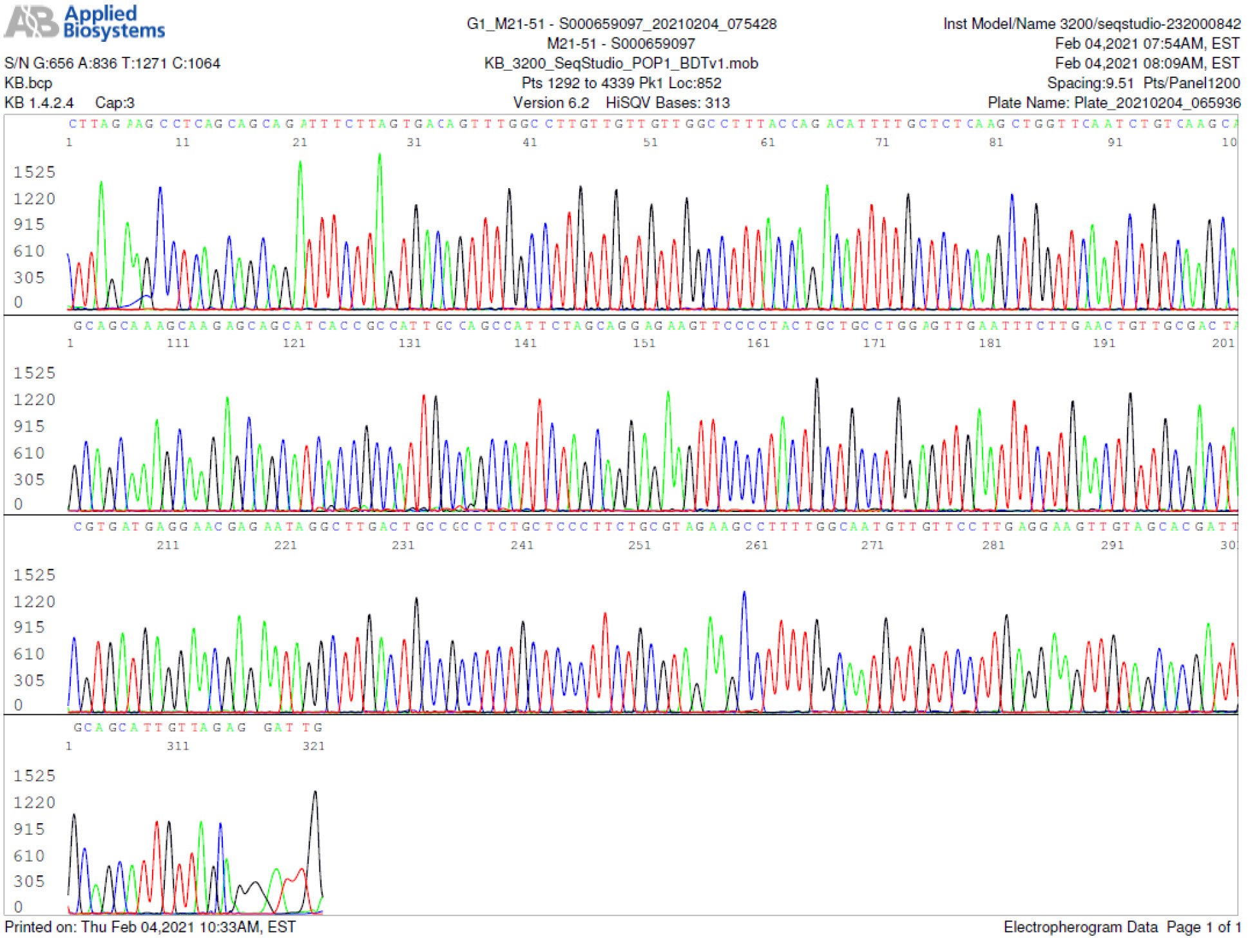

M21-52 Positive bi-directional sequencing and BLAST with 1 single nucleotide mutation typed in red
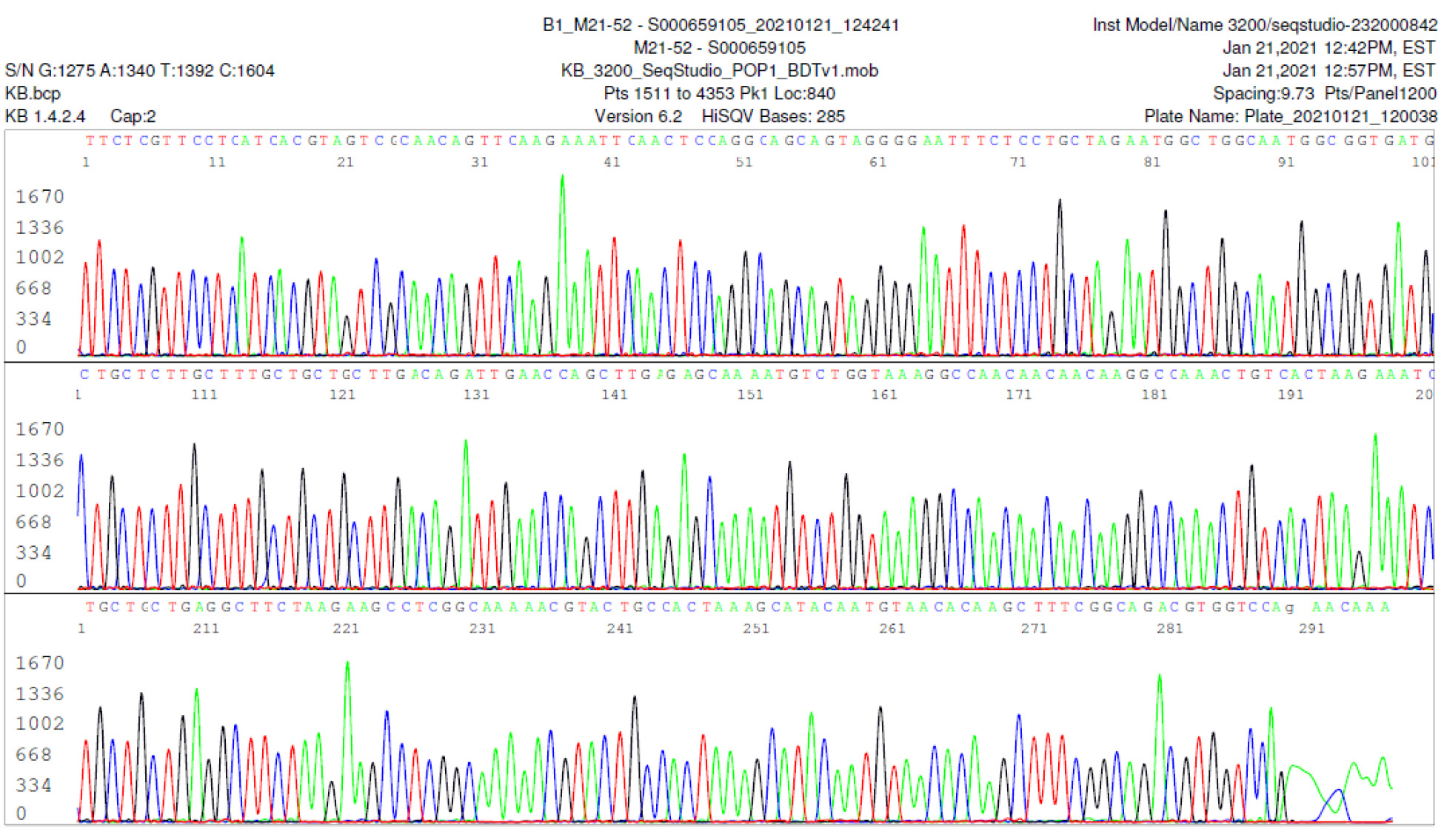
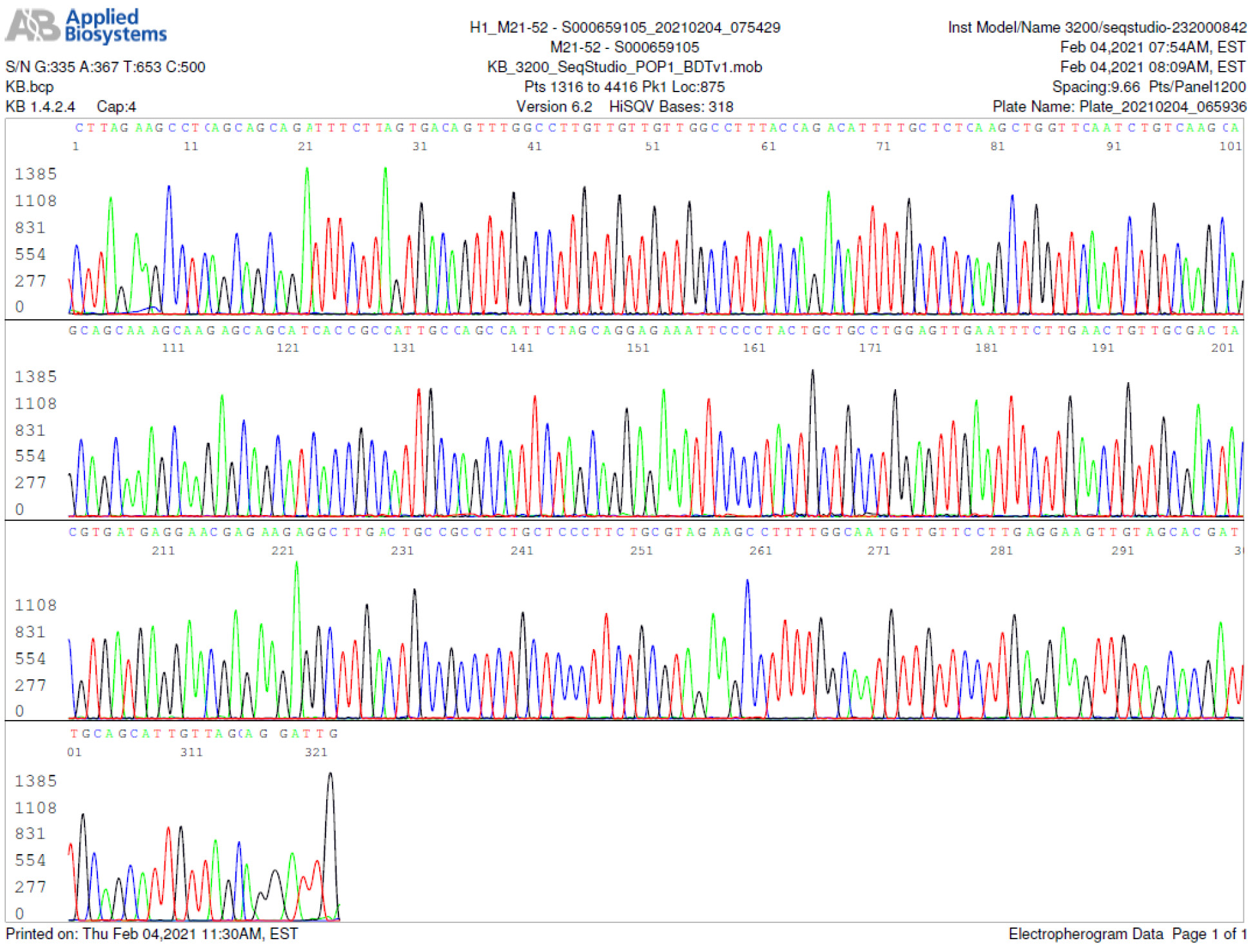
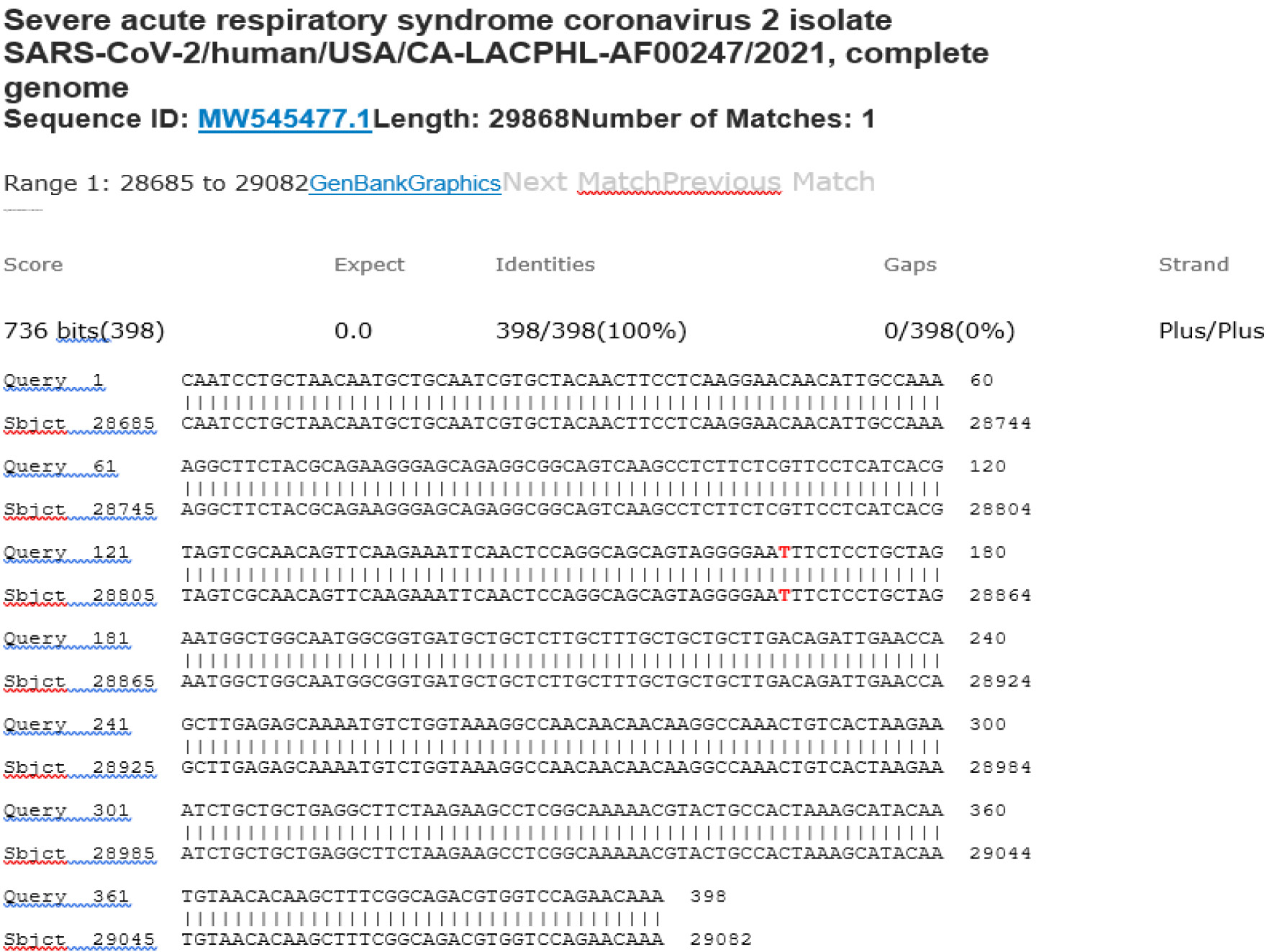
M21-53 Negative sequencing
M21-54 Positive bi-directional sequencing and BLAST with 1 single nucleotide mutation typed in red
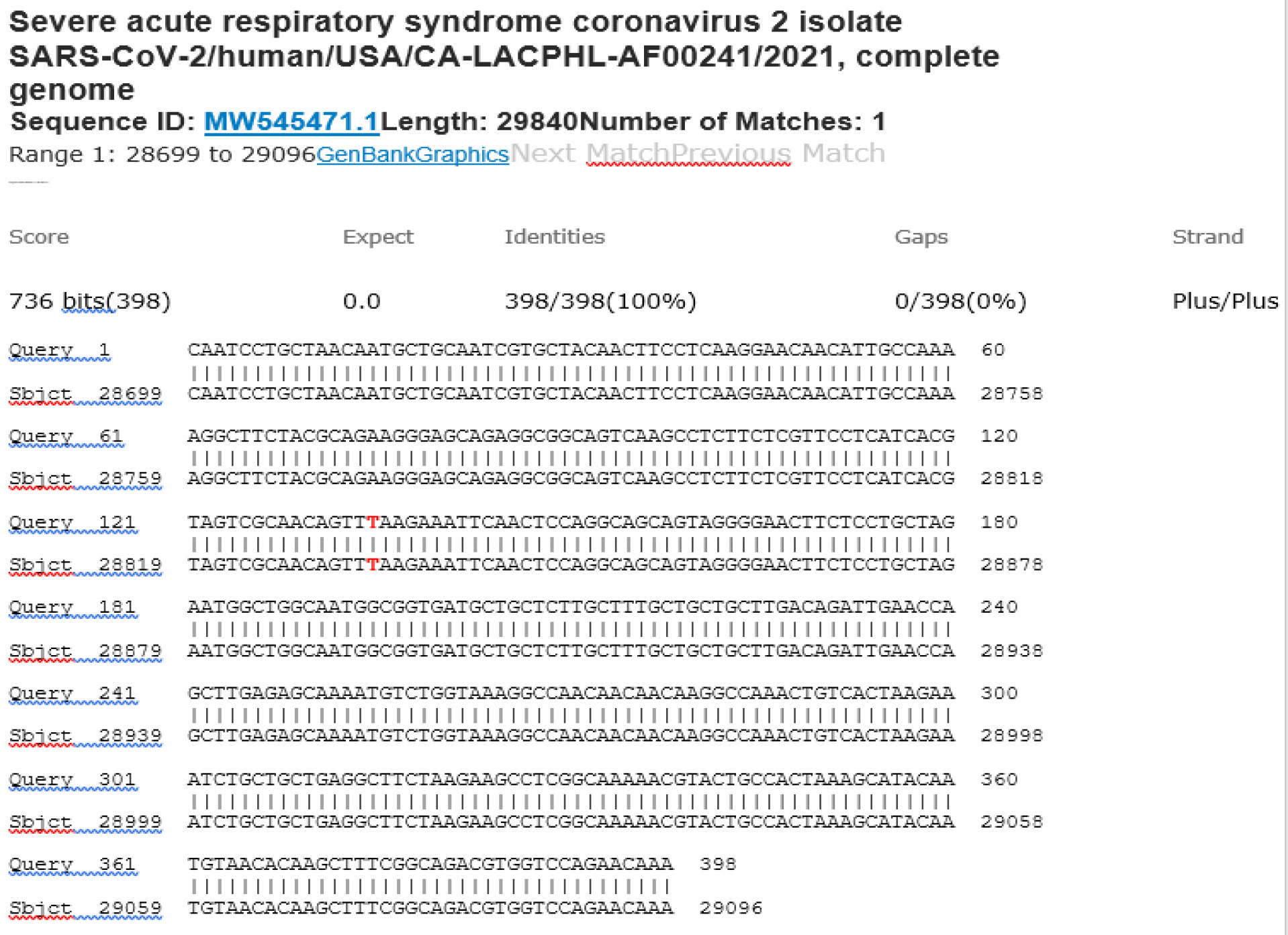
M21-55 Negative sequencing
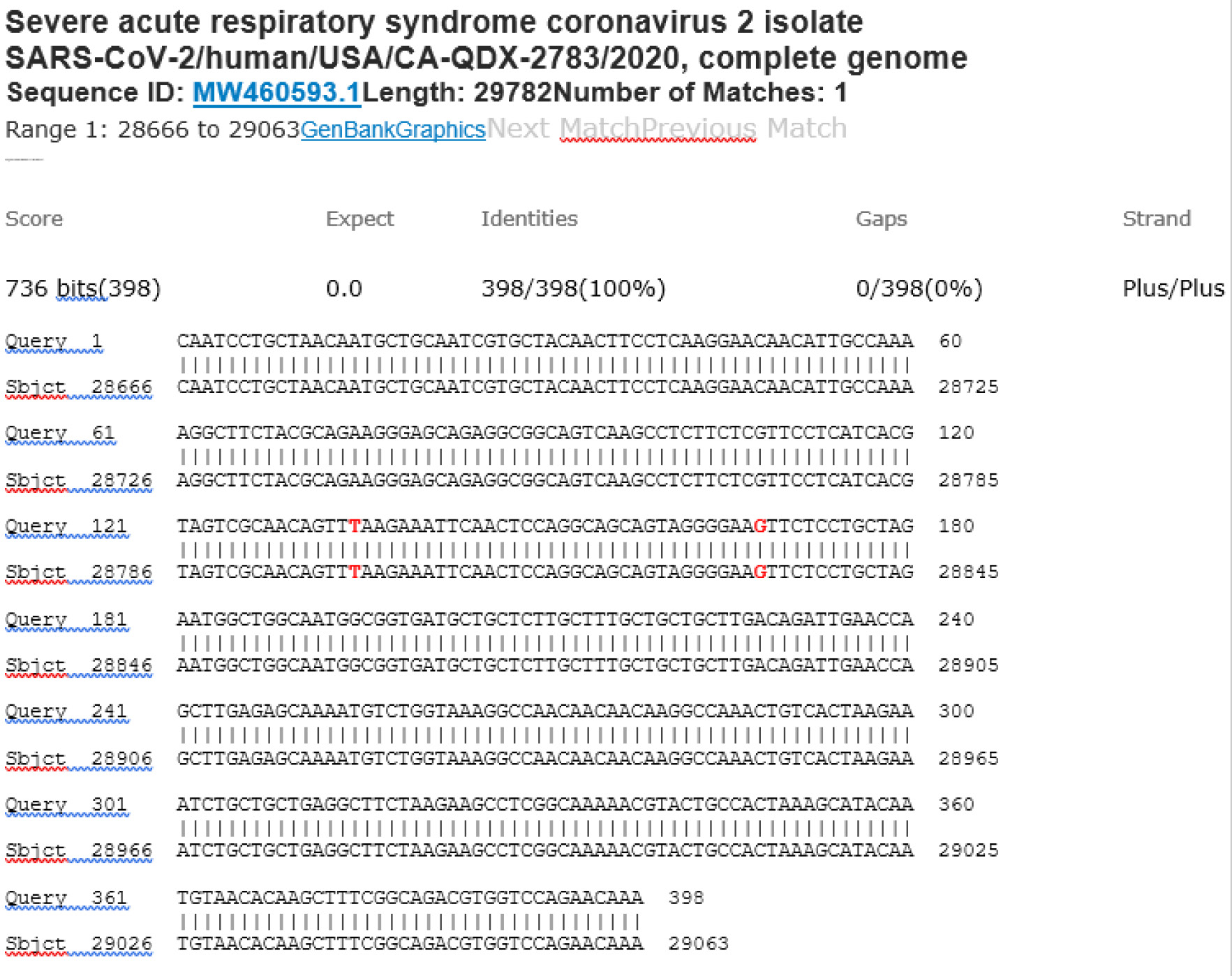
M21-56 Positive bi-directional sequencing and BLAST with 2 single nucleotide mutations typed in red
M21-57 Negative sequencing
M21-58 Positive bi-directional sequencing and BLAST with 1 single nucleotide mutation typed in red
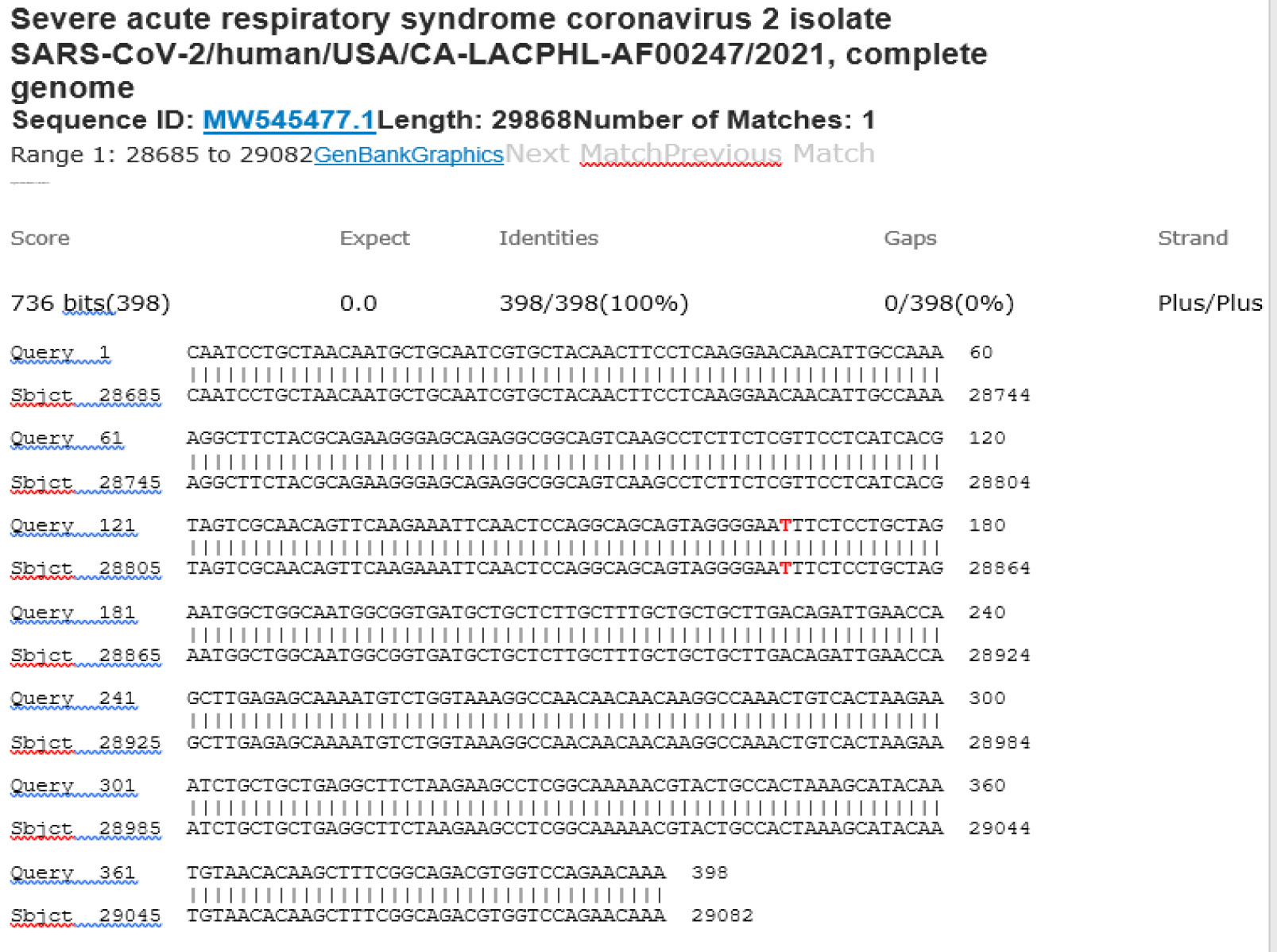
M21-59 Positive bi-directional sequencing and two BLAST reports showing a mutant with 1 single nucleotide C >T mutation co-existent with its wildtype parental virus
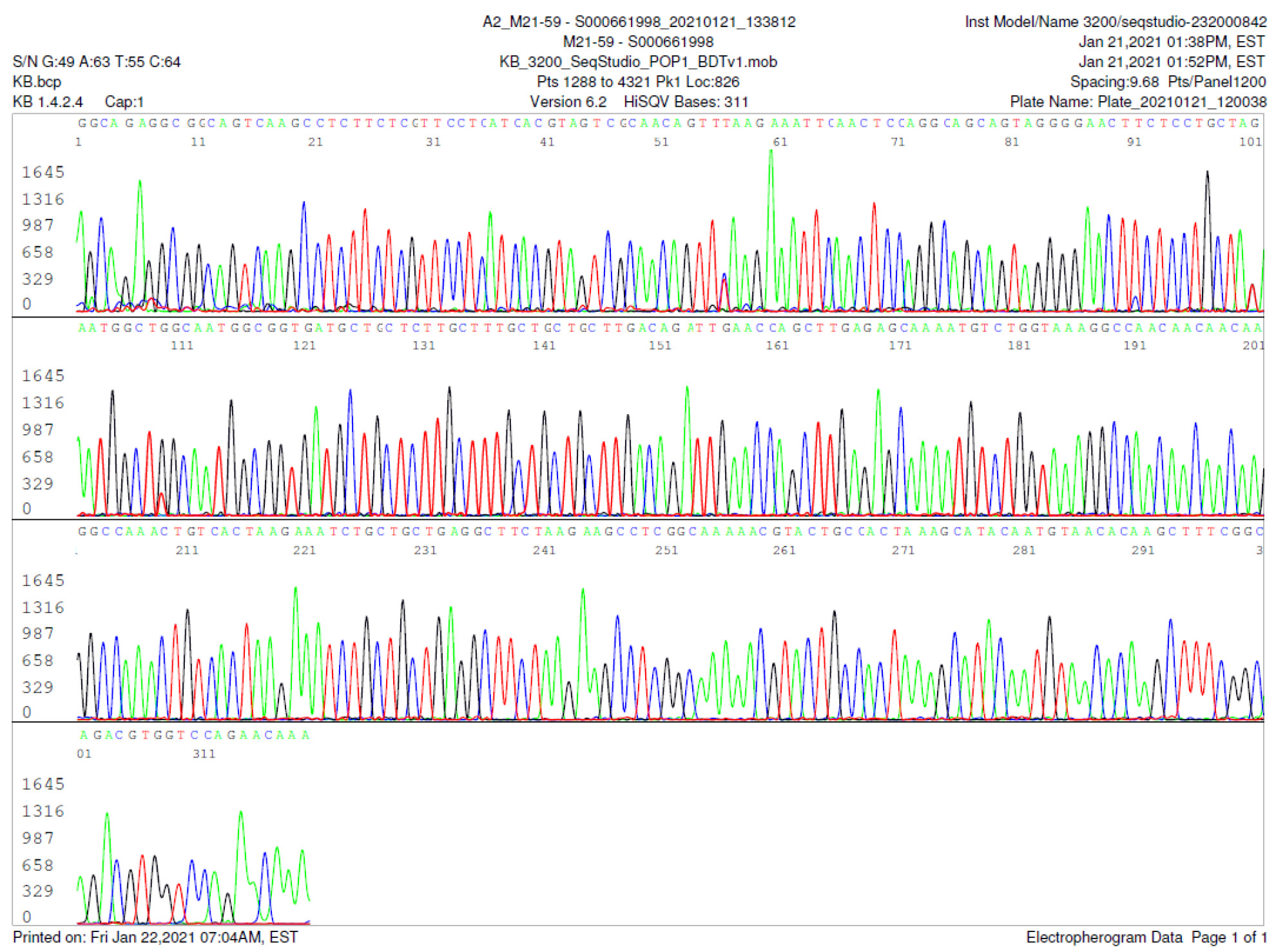
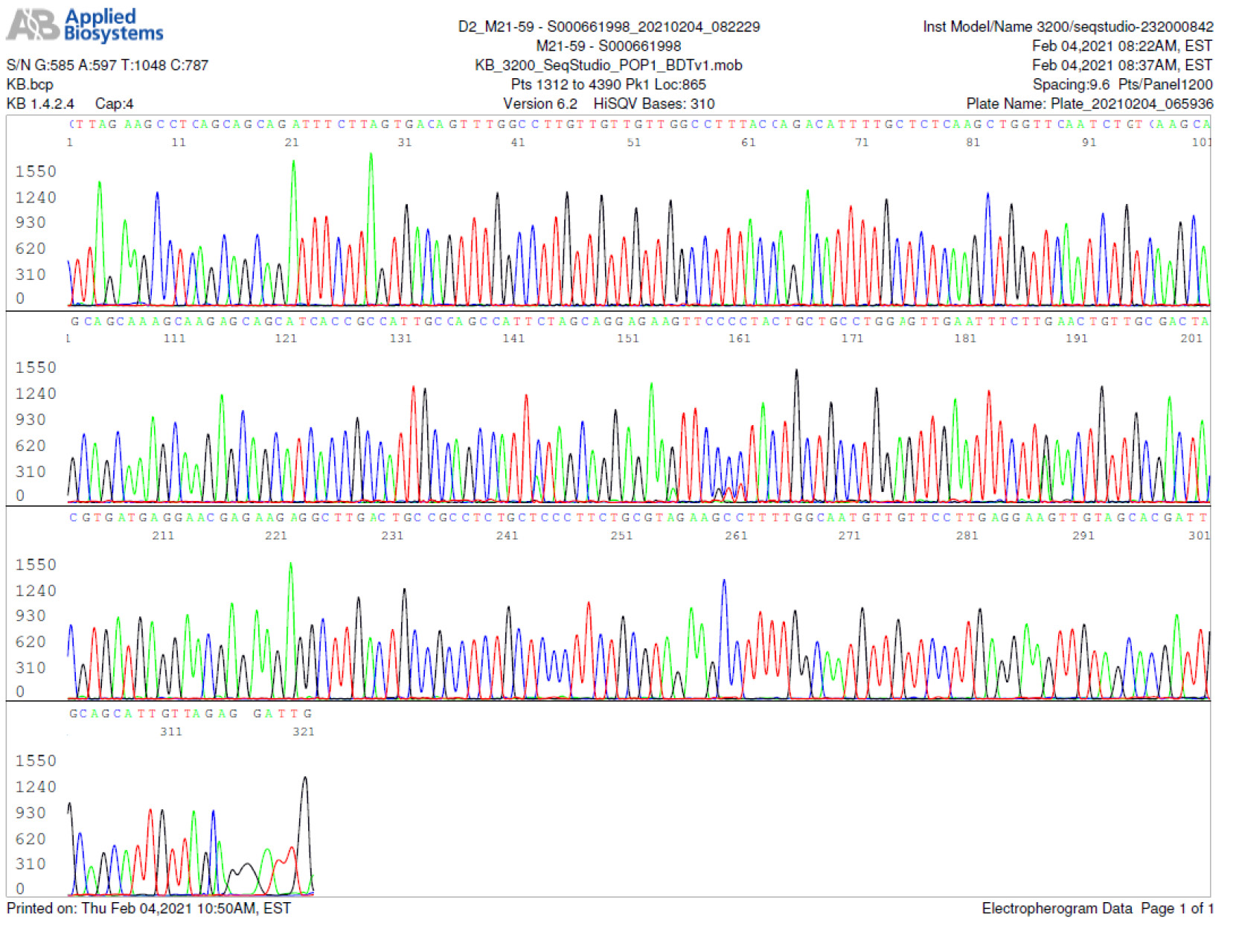
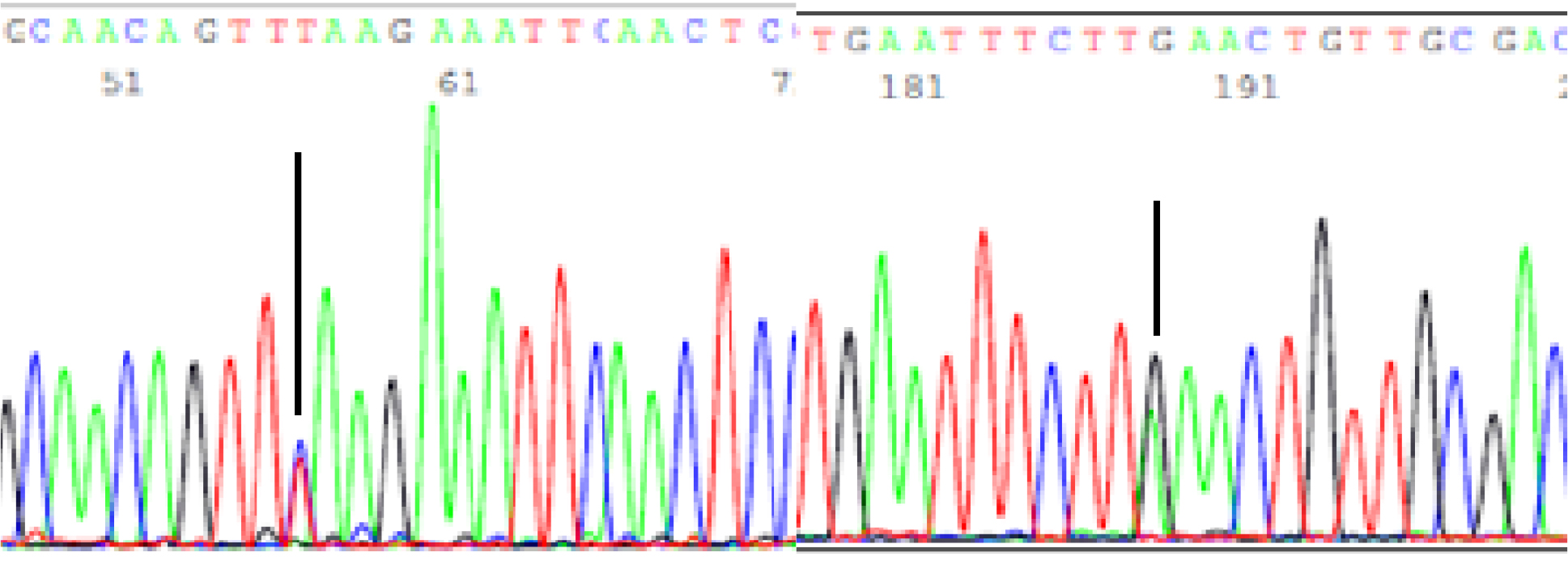
A mutant of SARS-CoV-2 with one base C > T mutation in this segment of N gene co-existent with its wildtype parental virus in this sample is demonstrated in the magnified bi-directional sequences excised from the electropherograms presented above. The superimposed T/C and G/A base peaks are indicated by a vertical bar. The double peak was read by the computer as T or G, depending on the strand (or direction) of the DNA template being sequenced.

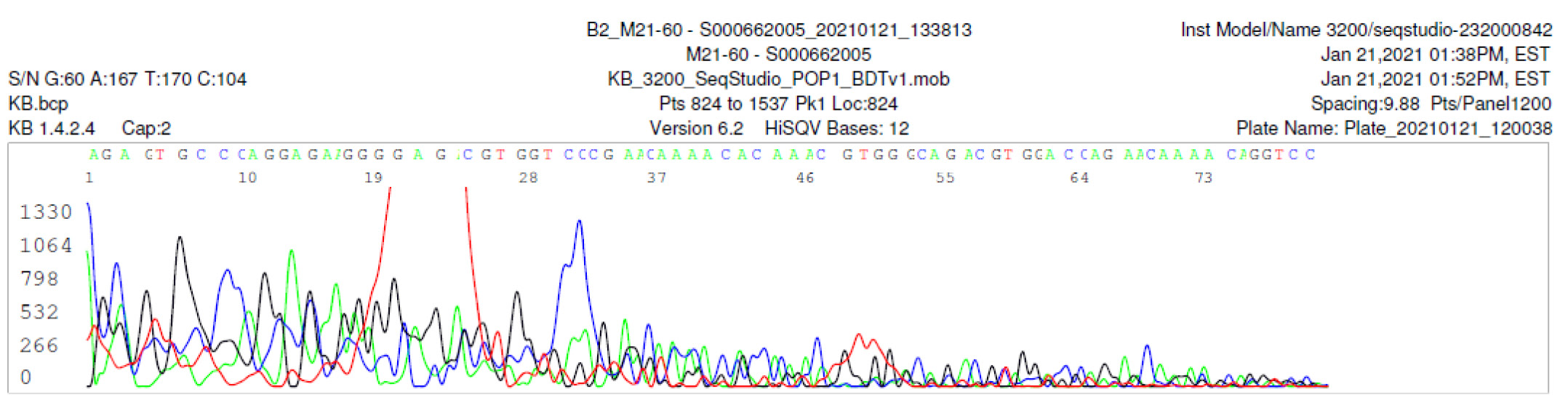
M21-60 Negative sequencing
Summary of single nucleotide mutations in partial SARS-CoV-2 N gene sequencing
All 16 positive samples showed mutations (typed in red) within a 79-base stretch of nucleic acid corresponding to positions 28821 to 28899 of the SARS-CoV-2 Wuhan-Hu-1 GenBank reference sequence (NC_045512.2)
